
Abstract
Kelloggia chinensis Franch. is an herbal plant species endemic to East Asia. Its complete plastid genome sequence is 155, 665 bp in length, with a large single-copy (LSC) region of 85, 788 bp, a small single-copy (SSC) region of 16, 977 bp, and a pair of inverted repeat regions (IRs) of 26, 450 bp. The whole plastid genome contains 132 genes, including 87 protein-coding genes, 37 tRNA genes, and 8 rRNA genes. The overall GC content of K. chinensis plastid genome is 37.1%. K. chinesis is evolutionarily close to tribe Rubieae according to the Maximum likelihood phylogenetic analysis based on 12 taxa.
Kelloggia Torrey ex Bentham is a small genus of Rubiaceae. It consists of only two species: Kelloggia chinensis Franch. from East Asia (Dempster Citation1975) and Kelloggia galioides Torrey from the western United States (Bentham and Hooker Citation1873). This genus shows a disjunction between eastern Asia and western North America. K. chinensis is a perennial herb occurs in alpine meadows or forest clearances at above 3, 000 m in East Asia (Chen and Ehrendorfer Citation2011). In the present study, we reported the first complete plastid genome of K. chinensis and performed the phylogenetic analysis with other related species within the subfamily of Rubioideae based on the plastid genome sequences.
Fresh leaves were collected from Daochen county, Sichuan province of China (99°42′55.03″E, 29°02′47.64″N). A specimen was deposited at the Herbarium of Yunnan Normal University (YNUB, [email protected]) under the voucher number YangLE649. Total genomic DNA was extracted from leaf tissue with an improved 4 x CTAB method (Doyle and Doyle Citation1987). Illumina paired- end (PE) library was constructed, and high-throughput genome sequencing was performed on the Illumina HiSeq X Ten platform. The GetOrganelle v1.7.0 (Jin et al. Citation2020) and the PGA (Qu et al. Citation2019) were used to assemble and annotate the chloroplast genome, respectively, with the whole plastid genome sequences of Galium mollugo L. (KY562588) as reference. The newly annotated complete plastid genome was submitted to GenBank (accession number OK236359).
The complete plastid genome of K. chinensis is 155, 665 bp in length, containing a pair of inverted repeats (IRs) of 26, 450 bp, a large single-copy (LSC) region of 85,788bp and a small single-copy (SSC) region of 16, 977 bp. The overall GC content of this genome is 37.1% (LSC, 37.1%; SSC, 31.3%; IRs, 42.9%). The whole plastid genome contains 132 genes, including 87 protein-coding genes, 37 tRNA genes, and 8 rRNA genes. Among the annotated genes 16 genes (atpF, ndhA, ndhB, rpl2, rpoC1, rps12, rps16, petB, petD, trnA-UGC, trnG-GCC, trnI-CAU, trnI-GAU, trnK-UUU, trnL-UAA, trnV-UAC) comprise one intron, and two introns are contained in clpP and ycf3.
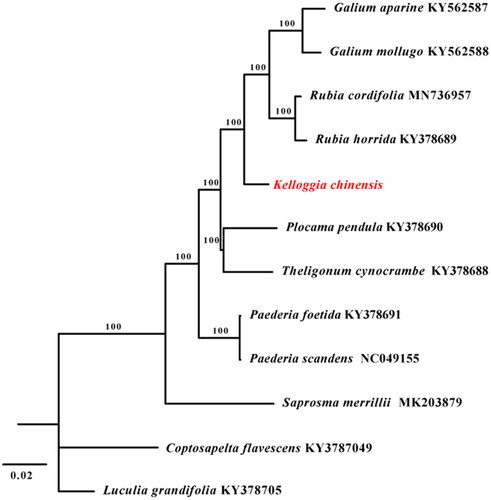
To clarify the phylogenetic position of K. chinensis, the complete plastid genomes of 10 species were selected within the subfamily of Rubioideae. Another two species from close subfamily were chosen as outgroups. The plastomes of the 12 accessions were aligned using MAFFT (Katoh and Standley Citation2013). A maximum likelihood analysis was performed using RAxML (Stamatakis Citation2006) software with GTR + G model and using the rapid bootstrap with 1000 replicates. The tribe of Rubieae is supported as a monophyly clade by the phylogenetic analysis, and K. chinensis is evolutionarily close to trirbe Rubieae and it is the sister group of this tribe (). This report provided a valuable resource for the future studies in Kelloggia and related taxa.
Figure 1. The maximum likelihood (ML) tree based on complete plastid genome sequences from 10 species within the subfamily of Rubioideae. Luculia grandifolia (KY378705) and Coptosapelta flavescens (KY3787049) were used as outgroups. The bootstrap support values with 1000 replicates are shown at each node.
Acknowledgements
We are grateful to Dr. Zhe Chen and Jun-Chu Peng for their help for data analysis.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at [https://www.ncbi.nlm.nih.gov] (https://www.ncbi.nlm.nih.gov/) under the accession no. OK236359. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA776541, SRR16643418, and SAMN22794791 respectively.
Additional information
Funding
References
- Bentham G, Hooker JD. 1873. Genera plantarum. Vol. 2. London, UK: Reeve.
- Chen T, Ehrendorfer F. 2011. Kelloggia Torrey ex Bentham & J. D. Hooker. In: Wu CY, Raven PH, Hong DY, editors. Flora of China. Vol. 19. Beijing & St. Louis: Science Press and Missouri Botanical Garden Press; p. 183–184.
- Dempster LT. 1975. An Asian Kelloggia (Rubiaceae). Madrono. 23:100–101.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Jin JJ, Yu WB, Yang JB, Song Y, de Pamphilis CW, Yi TS, Li DZ. 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):241.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Qu XJ, Moore MJ, Li DZ, Yi TS. 2019. PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods. 15:50.
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 22(21):2688–2690.