
Abstract
Leptinotarsa decemlineata Say 1824, an invasive and globally devastating beetle, inflicts great damage to potato crops worldwide. The complete mitogenome of L. decemlineata is described in this study. It is a 16,741 bp long circular DNA molecule with a high A + T content of 76.9%, containing a typical 37 gene pattern. All PCGs (protein-coding genes) initiate with typical ATN codons. Most PCGs use TAN as a stop codon, whereas ND4 and COX3 use the incomplete codon TA as the stop codon. The lengths of rrnL and rrnS genes are 1,337 bp and 811 bp, respectively. All 22 tRNAs ranged from 62 to 77 bp. Phylogenetic analysis of Chrysomelidae indicated that L. decemlineata clusteres with three other Chrysomelinae species, which is consistent with previous analyses.
The Colorado potato beetle, Leptinotarsa decemlineata Say 1824 (Coleoptera, Chrysomelidae), is one of the most destructive insect pests of potato (Cingel et al. Citation2016). Native to North America, it has now spread to and invaded countries worldwide (Hare Citation1990; Alyokhin Citation2009). The larvae and adults feed on foliage and tubers, causing severe damage to the plants (Alyokhin Citation2009). In 2017, adult specimens of L. decemlineata were collected from No.3 Donghuan Road, Gongliu County, Ili Kazak Autonomous Prefecture, Xinjiang Uygur Autonomous Region, China (82.3226°E, 43.4721°N). Voucher specimens were preserved in 99.7% ethanol and deposited in the Insect Collection at the College of Plant Protection, Hunan Agricultural University (please contact Yu-Sheng Wang, email: [email protected]) under the voucher number cpb2017083101. The genomic DNA was extracted from a single specimen using the DNeasy Tissue kit (Qiagen, Germany). The mitogenome was sequenced using the Illumina Hiseq X platform (Macrogen Inc., South Korea) and assembled by SPAdes v3.11.1 (Bankevich et al. Citation2012). MitoZ (Meng et al. Citation2019) software was used to annotate the mitogenome with reference to other Chrysomelinae species (MF563962, MK049855, MF198406).
The complete mitogenome of L. decemlineata (GenBank accession number MZ189364) is a 16,741 bp long circular molecule, comprised 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNAs), 2 ribosomal RNA genes (rrnL and rrnS), and one control region. Nine PCGs and 14 tRNAs were encoded on the H-strand, whereas the others were located on the L-strand. The nucleotide composition of the L. decemlineata mitogenome is significantly biased (39.4% A, 37.5% T, 14.1% G, and 8.9% C), with an overall A + T content of 76.9%. Meanwhile, the mitogenome presented a positive AT-skew and GC-skew (0.025 and 0.226, respectively). The gene arrangement is identical with the ancestral gene order of insects (Boore Citation1999) and Chrysomelinae species (Gómez-Rodríguez et al. Citation2015), but some Chrysomelinae species have translocations of rrnL and rrnS (Nie et al. Citation2020). All 13 PCGs initiate with typical ATN codons: three (COX1, ND5, and ND6) with ATT, four (ATP6, COX3, ND1, and ND4) with ATG, five (COX2, CYTB, ND2, ND3, and ND4L) with ATA, and one (ATP8) with ATC. ATP6, ATP8, COX1, COX2, CYTB, ND2, ND3, ND4L, and ND6 ended by TAA as the stop codon. ND1 and ND5 ended by TAG as the stop codon. Only ND4 and COX3 use the incomplete codon TA as their stop codon. The rrnL gene is 1,337 bp long with an A + T content of 82.1%, and the rrnS gene is 811 bp long with an A + T content of 80.3%, as is found in most insect mitogenomes. The control region, located between the rrnS and trnI genes, is 2,109 bp long with a remarkably high A + T content (80.5%). All 22 tRNAs range from 62 bp (trnL) to 77 bp (trnI), comprising a total length of 1,435 bp. Gene overlaps are found at 21 gene junctions and account for a total length of 177 bp, ranging from 1 to 38 bp long. The longest 38 bp overlap was located between rrnL and trnL1. A total of 86 bp intergenic spacer regions are present in nine positions, ranging from 1 to 21 bp long. The largest spacer sequence of 21 bp resided between trnL2 and COX2.
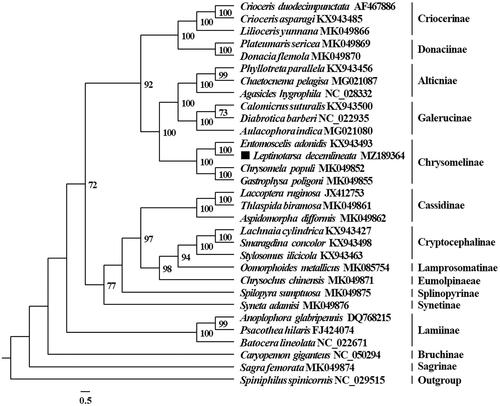
Phylogenetic analysis was performed with the concatenated nucleotide sequences of 13 PCGs genes from 30 Chrysomelidae species and Spiniphilus spinicornis (Vesperidae, as the outgroup). The 13 PCGs were partitioned using PartitionFinder2 (Lanfear et al. Citation2017). The phylogeny of Chrysomelidae was reconstructed using IQ-TREE (Nguyen et al. Citation2015) with the Maximum likelihood method. The ML phylogenetic tree supported that L. decemlineata clustered with three other Chrysomelinae species, and produced a Chrysomelidae phylogeny () which was consistent with previous analyses (Nie et al. Citation2020).
Figure 1. Maximum likelihood (ML) phylogenetic tree of 30 species of Chrysomelidae based on partitioned protein-coding sequences. ■ indicates the beetle determined in this study. The nodal numbers indicate the ML bootstrap support values (>70%).
Ethical approval
This study has been granted an exemption by the ethics committee of Hunan Agricultural University. Specific permission is not needed, because no relevant animals were involved.
Author contributions
TMD and YSW conceived and designed the study. All authors (TMD, HT, XL, GFZ, and YSW) analyzed and interpreted the results. TMD and YSW drafted the manuscript. All authors critically revised it for intellectual content and approved the final version to be published. All authors agree to be accountable for all aspects of the work.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The genome sequence data that supports the findings of this study are openly available in GenBank of NCBI at [https://www.ncbi.nlm.nih.gov] under the accession no. MZ189364. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA752722, SRR15367804, and SAMN20606172, respectively.
Additional information
Funding
References
- Alyokhin A. 2009. Colorado potato beetle management on potatoes: current challenges and future prospects. Fruit Veg Cereal Sci Biotech. 3(Suppl 1):10–19.
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Boore JL. 1999. Animal mitochondrial genomes. Nucleic Acids Res. 27(8):1767–1780.
- Cingel A, Savić J, Lazarević J, Ćosić T, Raspor M, Smigocki A, Ninković S. 2016. Extraordinary adaptive plasticity of Colorado potato beetle: ‘tenstriped spearman’ in the era of biotechnological warfare. IJMS. 17(9):1538.
- Gómez‐Rodríguez C, Crampton‐Platt A, Timmermans MJTN, Baselga A, Vogler AP. 2015. Validating the power of mitochondrial metagenomics for community ecology and phylogenetics of complex assemblages. Methods Ecol Evol. 6(8):883–894.
- Hare JD. 1990. Ecology and management of the Colorado potato beetle. Annu Rev Entomol. 35(1):81–100.
- Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. 2017. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol. 34(3):772–773.
- Meng GL, Li YY, Yang CT, Liu SL. 2019. MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 47(11):e63.
- Nguyen LT, Schmidt HA, Haeseler AV, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Nie RE, Andújar C, Gómez-Rodríguez C, Bai M, Xue HJ, Tang M, Yang CT, Tang P, Yang XK, Vogler AP. 2020. The phylogeny of leaf beetles (Chrysomelidae) inferred from mitochondrial genomes. Syst Entomol. 45(1):188–204.