
Abstract
Achnatherum pekinense belongs to Poaceae. The complete chloroplast genome of A. pekinense was reported in this study. The chloroplast genome was 137,837 bp in size with a canonical quadripartite structure, including two inverted repeat regions (IR) of 21,635 bp for each, a large single-copy (LSC) region of 81,787 bp in length, and a small single-copy (SSC) region of 12,780 bp in length. The overall guanine-cytosine (GC) content of this chloroplast genome was 38.8%, and the corresponding values of the LSC, SSC, and IR regions were 36.9%, 33.1%, and 44.1%, respectively. A total of 113 unique genes were annotated in this chloroplast genome, including four rRNA genes, 31 tRNA genes, and 78 protein-coding genes. The phylogenetic analysis showed that A. pekinense was clustered with A. inebrians.
Achnatherum pekinense [(Hance) Ohwi 1877] is a perennial herb of Gramineae, which is widely distributed on hillsides, grasslands, forests, beaches, and roadsides in northern China at an altitude of 350–1500 meters. At present, the study of A. pekinense mainly focused on leaf epidermal structure and powdery mildew (Chu and Yang Citation1991; Chen et al. Citation2018). From the perspective of genome, there are few studies on Achnatherum species. To date, Achnatherum chloroplast genomes are available for only two representatives, A. splendens and A. inebrians (Li et al. Citation2019; Wei et al. Citation2021). There are still many questions related to its phylogeny and species identification on Achnatherum. In this study, the chloroplast genome of A. pekinense was reported and its phylogenetic position was determined, which will contribute to the study of Achnatherum.
Fresh leaves were collected from Shandong Forest and Grass Germplasm Resources Center (Shandong, China 36°37′33.58″N, 117°9′58.97″E). The voucher specimen was deposited at College of Life Sciences, Shandong Normal University (Shou-Jin Fan, e-mail: [email protected]) under the voucher number SD469. Total genomic DNA was extracted by using the modified CTAB method (Doyle and Doyle Citation1987), and was sequenced by the Novaseq platform at Novogene (Beijing, China). The chloroplast genome assembly was performed with Getorganelle (Jin et al. Citation2020). The annotation of the chloroplast genome was performed with Plastid Genome Annotator (PGA) (Qu et al. Citation2019), and then manually corrected with Geneious v9.1.4 (Kearse et al. Citation2012). The sequence of complete chloroplast genome has been submitted to GenBank under accession number MZ680617.
The complete chloroplast genome of A. pekinense was 137,837 bp in length. The overall guanine-cytosine (GC) content was 38.8%. This chloroplast genome contained a total of 113 unique genes, including 78 protein-coding genes (PCGs), 31 transfer RNA genes (tRNAs), and four ribosomal RNA genes (rRNAs). A total of 10 PCGs contained introns, of which eight PCGs (atpF, ndhA, ndhB, petB, petD, rpl16, rpl2, and rps16) contained one intron and two PCGs (rps12 and ycf3) contained two introns.
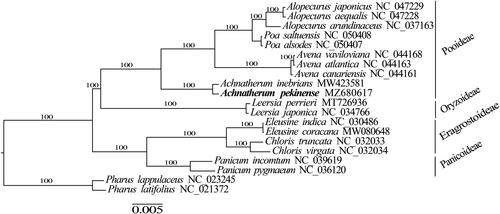
A maximum-likelihood (ML) tree was reconstructed to determine the phylogenetic relationships of A. pekinense by using RAxML v8.2.10 (Stamatakis Citation2014), with tree robustness assessment with 1000 rapid bootstrap replicates, and the substitution model was GTRGAMMA. Alignment of 78 shared PCGs was conducted by using MAFFT v7.313 (Katoh and Standley Citation2013). ML phylogenetic analysis showed that A. pekinense was sister to A. inebrians ().
Figure 1. A maximum-likelihood (ML) tree inferred from 78 protein-coding genes is shown. Pharus lappulaceus and Pharus latifolius are used as outgroup. The numbers on branches are bootstrap support values.
Authors contributions
Conceiving and designing, S.-Q.X. and S.-J.F.; performing and analyzing data, S.-Q.X., B.-Q.T, B.Z.; writing – original draft preparation, S.-Q.X.; writing – review and editing, S.-Q.X.; supervision, B.-Q.T, B.Z., and S.-J.F.; All authors have read and agreed to the published version of the manuscript.
Acknowledgements
Ethical statement: The plant was collected in the wild, and there was no relevant ethical conflict. No ethical approval required.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov/ under the accession no. MZ680617. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA751256, SRR15316867, and SAMN20513650, respectively.
Additional information
Funding
References
- Chen YF, Yu SR, Du Y, Kong LJ. 2018. Investigation on the correlation between cereal powdery mildew and overwintering of wheat powdery mildew in Northern Jiangsu. B Agric Sci Technol Iran.12(3): 214–216.
- Chu QG, Yang YC. 1991. Studies on the leaf epidermal characters and its taxonomic significance of the genus Achnatherum in China. J Laiyang Agric Coll. 8(3):159–165.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Jin JJ, Yu WB, Yang JB, Song Y, dePamphilis CW, Yi TS, Li DZ. 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):241.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Li ZG, Jia GL, Ni XL. 2019. The complete chloroplast genome sequence of Achnatherum splendens (Pooideae), a high-quality forage grass in Northern China. Mitochondrial DNA B. 4(1):1841–1843.
- Qu XJ, Moore MJ, Li DZ, Yi TS. 2019. PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods. 15(1):50.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Wei X, Li X, Chen T, Chen Z, Jin Y, Malik K, Li C. 2021. Complete chloroplast genomes of Achnatherum inebrians and comparative analyses with related species from Poaceae. FEBS Open Bio. 11(6):1704–1718.