
Abstract
Urtica angustifolia Fisch. ex Hornem. is an important Chinese medicine. Here, the complete chloroplast genome of U. angustifolia was assembled and characterized. The length of the chloroplast genome was 146,679 bp with the typical quadripartite structure, containing two inverted repeats (IRs) of 24,595 bp separated by a large single-copy (LSC) region of 79,820 bp and a small single-copy (SSC) region of 17,669 bp. The whole chloroplast genome of U. angustifolia contains 111 genes, including 77 protein-coding genes, 30 tRNA genes, and 4 rRNA genes. Nucleotide variability analysis identified three hotspot regions (trnK-rps16, ndhF-rps32, and ycf1b) for genomic divergence and 52 simple sequence repeats. Phylogenetic analysis based on the complete chloroplast genomes exhibited that U. angustifolia formed a clade with Urtica lobatifolia and Hesperocnide tenella.
Urtica angustifolia Fisch. ex Hornem., belonging to Urticaceae, is mainly distributed in northern China, eastern Siberia, Russia, Mongolia, Korea and Japan. Urtica angustifolia is a traditional medicinal material (Zhang and Li Citation2012). However, the genetic information was less available. The chloroplast genome sequences provide an effective genetic resource for resolving complex evolutionary relationships, assessing population genetics, and identifying species (Dong, Liu, et al. Citation2021; Dong, Sun, et al. Citation2021; Wang, et al. Citation2021; Dong, Liu, et al. Citation2022). In the current study, we sequenced the complete chloroplast genome of U. angustifolia using Illumina Hiseq X ten platform.
Fresh and healthy herb material of U. angustifolia was collected from Maorshan mountain, Shangzhi, Heilongjiang, China (45°17′55″–127°31′49″). The collection of plant material was in accordance with local regulations and obtain the permission of local authorities. The voucher specimen was deposited at the herbarium of Jiangxi Agricultural University under the voucher number of ENC850487 (Mu Liu, [email protected]). The total genomic DNA was extracted from the fresh leaves with the modified CTAB method (Li et al. Citation2013). Genomic DNA was fragmented to construct a shotgun library with an insert size of 350 bp. The library was sequenced using the Illumina HiSeq X-ten platform and approximately 4 Gb data was generated from the sequencing library. Raw data were qualified by using Trimmomatic (Bolger et al. Citation2014) and the chloroplast genome was assembled with GetOrganelle (Jin et al. Citation2019). The complete chloroplast genome of U. angustifolia was annotated with Plann (Huang and Cronk Citation2015) using U. lobatifolia (Urticaceae, GenBank accession number: MW246155) as a reference. The annotated chloroplast genome of U. angustifolia has been deposited into GenBank with the accession number of MZ145046.
The length of the U. angustifolia chloroplast genome is 146,679 bp including two inverted repeats (IR, 24,595 bp), a large single-copy region (LSC, 79,820 bp) and a small single-copy region (SSC, 17,669 bp). The GC content of the genome is 36.6%. The U. angustifolia chloroplast genome is predicted to contain 111 unique genes, including 77 protein-coding genes, 30 tRNA genes and 4 rRNA genes. Among these genes, 12 genes (atpF, ndhA, ndhB, petB, petD, rpoc1, rpl16, trnA-UGC, trnI-GAU, trnK-UUU, trnL-UAA, and trnV-UAC) exhibit one intron and two genes (clpP and ycf3) contain two introns.
The Perl script MISA was used to identify microsatellites with the minimum numbers of repeats set to 10, 5, 4, 3, 3, and 3 for mono-, di-, tri-, tetra-, penta-, and hexanucleotides, respectively. The total number of SSRs identified in U. angustifolia chloroplast genome was 52. We calculated the nucleotide diversity (PI) using sliding window method (window size: 800 bp and step size: 200 bp) to identify the mutation hotspots in the chloroplast genome alignments of three Urtica samples and Hesperocnide tenella. Three variable regions (trnK-rps16, ndhF-rps32, and ycf1b) were found to be more variable with Pi values >0.05. We also identified 403 indels in the four aligned chloroplast genomes.
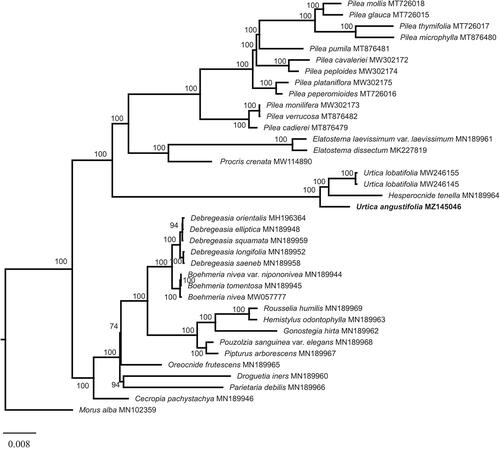
To resolve the phylogenetic position of U. angustifolia, a total of 36 chloroplast genome sequences (one outgroup from Moraceae) were downloaded from the NCBI database. Sequences were aligned using MAFFT (Katoh and Standley Citation2013) and the ambiguous alignment regions were trimmed using Gblocks 0.91 b (Castresana Citation2002). The maximum-likelihood (ML) phylogenetic tree was reconstructed using RAxML (Stamatakis Citation2014). ML analysis was performed using RAxML with 500 replications under the GTR + G model. The result showed that Urticaceae was divided into two clades and U. angustifolia formed a clade with Urtica lobatifolia and Hesperocnide tenella (). This result is consistent with the phylogenetic relationship based on several chloroplast genes, which also supported Hesperocnide tenella was within the genera Urtica (Wu et al. Citation2013, Citation2018). Our study will provide valuable insight into conservation genetics, taxonomy, and evolutionary histories for this particular species.
Figure 1. Phylogenetic tree of Urticaceae based on 37 complete chloroplast genome sequences. ML topology shown with ML bootstrap support value at each node.
Author contributions
Mu Liu and Lvshui Zhang designed the project. Mu Liu and Jinsen Lu collected samples and performed the experiment. Mu Liu, Jinsen Lu, and Baoyong Li analyzed the data. Mu Liu and Lvshui Zhang wrote and revised the paper. All authors have read and agreed to the published version of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at (https://www.ncbi.nlm.nih.gov/) under the accession no. MZ145046. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA749813, SRR15253676, and SAMN20422813, respectively.
Additional information
Funding
References
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30(15):2114–2120.
- Castresana J. 2002. GBLOCKS: selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. EMBL.
- Dong W, Liu Y, Li E, Xu C, Sun J, Li W, Zhou S, Zhang Z, Suo Z. 2022. Phylogenomics and biogeography of Catalpa (Bignoniaceae) reveal incomplete lineage sorting and three dispersal events. Mol Phylogenet Evol. 166:107330.
- Dong W, Liu Y, Xu C, Gao Y, Yuan Q, Suo Z, Zhang Z, Sun J. 2021. Chloroplast phylogenomic insights into the evolution of Distylium (Hamamelidaceae). BMC Genomics. 22(1):293.
- Dong W, Sun J, Liu Y, Xu C, Wang Y, Suo Z, Zhou S, Zhang Z, Wen J. 2021. Phylogenomic relationships and species identification of the olive genus Olea (Oleaceae). J Syst Evol.
- Huang DI, Cronk QCB. 2015. Plann: a command-line application for annotating plastome sequences. Appl Plant Sci. 3(8):1500026.
- Jin J-J, Yu W-B, Yang J-B, Song Y, dePamphilis CW, Yi T-S, Li D-Z. 2019. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. bioRxiv:256479.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Li J, Wang S, Jing Y, Wang L, Zhou S. 2013. A modified CTAB protocol for plant DNA extraction. Chin Bull Bot. 48(1):72–78.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Wang M, Wang X, Sun J, Wang Y, Ge Y, Dong W, Yuan Q, Huang L. 2021. Phylogenomic and evolutionary dynamics of inverted repeats across Angelica plastomes. BMC Plant Biol. 21(1):26.
- Wu Z-Y, Liu J, Provan J, Wang H, Chen C-J, Cadotte MW, Luo Y-H, Amorim BS, Li D-Z, Milne RI. 2018. Testing Darwin's transoceanic dispersal hypothesis for the inland nettle family (Urticaceae). Ecol Lett. 21(10):1515–1529.
- Wu Z-Y, Monro AK, Milne RI, Wang H, Yi T-S, Liu J, Li D-Z. 2013. Molecular phylogeny of the nettle family (Urticaceae) inferred from multiple loci of three genomes and extensive generic sampling. Mol Phylogenet Evol. 69(3):814–827.
- Zhang H-Y, Li Q. 2012. Optimization of ultrasonic-assisted extraction of alkaloids from whole herbs of Urtica angustifolia using response surface methodology. Food Sci. 12.