
Abstract
The complete mitochondrial genome (mitogenome) of Laothoe amurensis sinica (Rothschild & Jordan, 1903) was sequenced. The L. amurensis sinica mitogenome is circular, double-stranded, with length of 15,341 bp. Gene content, gene order and orientation are all typical of Sphingidae. Nucleotide composition is highly biased toward A + T nucleotides (79.2%). Most of 13 protein-coding genes (PCGs) initiate with the standard start codon of ATN and terminate with the typical stop codon TAA/TAG or incomplete T. Phylogenetic analyses based on the maximum-likelihood (ML) on the W-IQ-Tree web server showed that L. amurensis sinica have a close relationship to the lineage formed by Clanis bilineata and Leucophlebia lineata.
The order Lepidoptera is one of the largest insect orders, which includes greater than 160,000 described species that are classified into 45–48 superfamilies (Hao et al. Citation2012). As one of the most diverse families, Sphingidae contains 203 genera and 1348 species distributed worldwide. Despite this enormous species diversity, study on its mitochondrial genome is still sparse. The hawk moth, Laothoe amurensis sinica (Rothschild & Jordan, 1903) is the species of Sphingidae, which is endemic to China and Korea. However, the mitogenome sequence of L. amurensis sinica remains unknown so far. Therefore, complete mitochondrial genome of L. amurensis sinica was sequenced in this study in order to determine the mitochondrial genome of this species and to reconstruct a phylogenetic tree of Sphingidae.
Laothoe amurensis sinica was collected from the Dabie Mountain, Lu'an City, Anhui Province, China (31°13′08″N, 116°20′19″E) in May 2021 and deposited in the Entomological Museum, College of Life Sciences, Anhui Normal University (https://www.ahnu.edu.cn/, YX, Huang, [email protected]) under the accession no. DB202105225. All animal-related experiments were performed according to the protocols approved by the Institutional Animal Care and Use Committee of Anhui Normal University (Grant number AHNU-ET2021032). A whole genome shotgun (WGS) strategy was used with sequencing on the Illumina novaseq 6000 platform (Andrews Citation2020). The raw paired reads were quality-trimmed and assembled into the complete circular mitogenome in Novoplasty 2.7.2.
The complete mitogenome of L. amurensis sinica (GenBank accession number: MZ593601) is circular, double-stranded, and 15,341 bp long. The typical 37 mitochondrial genes (13 PCGs, 22 tRNAs, and 2 rRNAs) and an A + T-rich region are included. The overall base composition of the mitogenome was estimated to be A: 40.9%, T: 38.3%, C: 12.9%, and G: 7.9%. The composition of A, T, G, C nucleotides in the L. amurensis sinica mitogenome indicated a remarkably high AT content (79.2%) and a low GC content (20.8%). The heavy strand (H-strand) encoded more genes (9 PCGs and 14 tRNAs) than the light strand (L-strand) that encoded 4 PCGs, 8 tRNAs, and 2 rRNAs. A total of 13 PCGs were identified in L. amurensis sinica, with a length of 11,193 bp. The 13 PCGs of the mitogenome include 7 NADH dehydrogenase subunits, 3 cytochrome c oxidase subunits, 2 ATPase subunits, and one cytochrome b gene. Most of 13 PCGs start with ATN and stop with traditional TAA/TAG codons or incomplete T, which is similar to most other insect mitogenomes (Crozier and Crozier Citation1993; Korkmaz et al. Citation2015).
Laothoe amurensis sinica mitochondrial tRNA genes were scattered throughout the molecule, with 14 encoded by the H-strand, while the rest were encoded by the L-strand. There were two rRNAs in L. amurensis sinica with a total length of 2192 bp. The length of large ribosomal gene (rrnL) was 1383 bp, whereas the length of small ribosomal gene (rrnS) was only 809 bp.
To reconstruct the phylogenetic relationship among hawkmoths, the nucleotide sequences of the 13 PCGs were first aligned and then concatenated. A total of 27 taxa, with 24 Sphingidae and 2 outgroups from GenBank were sampled for phylogenetic analyses. Nucleotide sequences from each PCG were aligned by MAFFT (Katoh et al. Citation2005), and then concatenated the aligned sequences into a dataset. Subsequently, the nucleotide sequences of PCGs were analyzed using the maximum-likelihood (ML) on the W-IQ-Tree web server to reconstruct the phylogenetic relationship of L. amurensis sinica with other Sphingidae (Trifinopoulos et al. Citation2016).
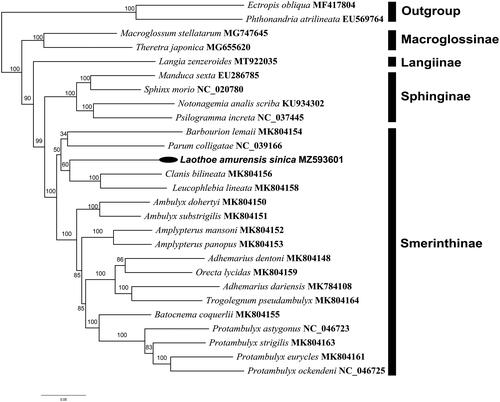
The result indicated that all the subfamilies formed monophyletic groups respectively, which was in accordance with our previous study (Wang et al. Citation2021). As shown in , the phylogenetic analyses showed that L. amurensis sinica have a close relationship to the lineage formed by Clanis bilineata and Leucophlebia lineata, which was well supported by the ML analyses.
Figure 1. Maximum likelihood phylogenetic tree for Sphingidae based on the nucleotide sequence data of 13 PCGs from Laothoe amurensis sinica and other 24 species belonging to four related subfamilies of Sphingidae. The number on each node indicates the values of ultrafast bootstrap (UFB) of 1000 replications.
Credit authorship statement
Yang Sun: the conception and design, analysis and interpretation of the data, the drafting of the paper, revising it critically for intellectual content. Jing Wang: the conception and design, analysis and interpretation of the data. Xu Wang: the conception and design, analysis and interpretation of the data, the drafting of the paper, revising it critically for intellectual content and the final approval of the version to be published. All authors agree to be accountable for all aspects of the work.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in GenBank at https://www.ncbi.nlm.nih.gov/genbank/, reference number MZ593601. The associated BioProject, Bio-Sample numbers, and SRA are PRJNA753228, SAMN20691822, and SRR15400582, respectively.
Additional information
Funding
References
- Andrews S. 2020. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- Crozier RH, Crozier YC. 1993. The mitochondrial genome of the honeybee Apis mellifera: complete sequence and genome organization. Genetics. 133(1):97–117.
- Hao J, Sun Q, Zhao H, Sun X, Gai Y, Yang Q. 2012. The complete mitochondrial genome of Ctenoptilum vasava (Lepidoptera: Hesperiidae: Pyrginae) and its phylogenetic implication. Comp Funct Genomics. 2012:328049–328013.
- Katoh K, Kuma KI, Toh H, Miyata T. 2005. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33(2):511–518.
- Korkmaz EM, Doğan Ö, Budak M, Başıbüyük HH. 2015. Two nearly complete mitogenomes of wheat stem borers, Cephus pygmeus (L.) and Cephus sareptanus Dovnar-Zapolskij (Hymenoptera: Cephidae): An unusual elongation of rrnS gene. Gene. 558(2):254–264.
- Trifinopoulos J, Nguyen LT, Haeseler AV, Minh BQ. 2016. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44(W1):W232–235.
- Wang X, Zhang H, Kitching I, Xu ZB, Huang YX. 2021. First mitogenome of subfamily Langiinae (Lepidoptera: Sphingidae) with its phylogenetic implications. Gene. 789:145667.