
Abstract
The benthic and tube-building polychaete worm Mesochaetopterus japonicus is abundantly present on the coast of the western Pacific. Here, we report the complete mitochondrial genome of M. japonicus, which is 19,326 bp in length and contains 13 protein-coding genes, 2 rRNA genes and 22 tRNA genes. All 37 genes are encoded on the heavy strand, and AT content is 70.17%. Phylogenetic analyses based on the M. japonicus mitogenome combined with previously published polychaete mitogenome data revealed that M. japonicus was closely related to Chaetopterus variopedatus and Phyllochaetopterus sp., all of which belong to Chaetopteridae. The mitochondrial genome of M. japonicus could provide useful molecular resources for further research on Polychaeta phylogeny and evolution.
Mesochaetopterus japonicus (Fujiwara Citation1934) is known to be a tube-dwelling polychaete worm and is characterized by three well-defined body regions: anterior, middle and posterior. The species is widely distributed on the coast of the western Pacific (Yang and Sun Citation1988; Nishi Citation1999; Nishi and Hsieh Citation2009) and belongs to the family Chaetopteridae. Chaetopteridae is a globally distributed clade of marine annelids that inhabit areas from intertidal to abyssal depths. There are 76 valid species in four currently recognized genera (Moore et al. Citation2017): Chaetopterus Cuvier, 1830, Mesochaetopterus Potts, 1914, Phyllochaetopterus Grube, 1863, and Spiochaetopterus Sars, 1856 (WoRMS).
The mitochondrial genome (mitogenome) contains abundant genetic information and has a relatively high evolutionary rate (Boore Citation1999; Barr et al. Citation2005; Hao et al. Citation2010), so it is extensively used for studying genetic diversity, phylogeny, molecular evolution, and phylogeography at various taxonomic levels (Gissi et al. Citation2008; Shen et al. Citation2017; Lee et al. Citation2019). However, compared to other polychaetes, information one the Chaetopterid mitogenome is limited, with an examination of the NCBI nr database in August 2021 revealing that only two complete mitogenomes have been published. Herein, we determined the complete mitogenome of the Chaetopterids M. japonicus collected from Qingdao (36°03′N, 120°20′E) and deposited in the Marine Biological Museum (Specimen ID: MBM304570, Collection Manger Dr Mei Yang, [email protected]), Institute of Oceanology, Chinese Academy of Sciences, Qingdao, China. Research and collection of animal material was conducted according to the guidelines provided by Science and Technology Ethics Committee of Chinese Academy of Sciences.
Genomic DNA was extracted from the body tissue using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany) and sequenced on the Illumina HiSeq 4000 platform (2 × 150 bp paired-end reads). The mitogenome of M. japonicus was de novo assembled by using SOAPdenovo 2.04 (Luo et al. Citation2012) and then annotated by the MITOS2 webserver (Bernt et al. Citation2013). Finally, the mitogenome was manually corrected.
The complete circular mitogenome of M. japonicus is 19,326 bp in length (GenBank Accession no. MZ921947) and contains the typical set of 13 protein-coding genes (PCGs), 22 tRNA genes, 2 rRNA genes, and 1 putative control region. All 37 genes are encoded on the heavy strand, whose nucleotide composition is 34.36% A, 18.70% C, 11.13% G, and 35.81% T, showing a higher content of A + T (70.17%) than G + C (29.83%). The codon ATG was the most popular start codon for atp6, atp8, cox2, nad3, nad4l, and nad6, and the start codon ATT was shared among cob, nad1, and nad5. In particularly, cox3 and nad4 begins with the codon ATC, nad2 begins with the codon ATA, and cox1 begins with the codon AGA. For stop codon usage, the cox1 and nad2 stop with the incomplete stop codon, while the other PCGs terminate with TAA/TAG. Twenty-two tRNA genes varied from 65 to 76 bp in length, and all of them could fold into the typical cloverleaf secondary structure.
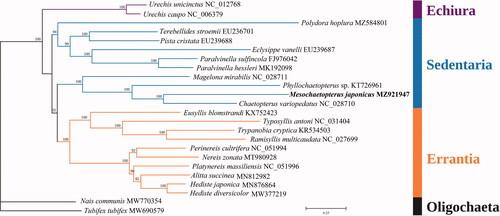
The phylogenetic tree was constructed based on 13 PCGs of M. japonicus and 21 other polychaetes from three main lineages, including Echiura, Errantia and Sedentaria. The oligochaetes Acanthobdella peledina and Tubifex tubifex were used as outgroups (). Relevant sequences were downloaded from the NCBI database and aligned by MAFFT (Katoh and Standley Citation2013) with the default parameters. Then phylogenetic relationships were inferred using the maximum-likelihood (ML) method with the GTR + F+R4 model by using IQ-TREE (Nguyen et al. Citation2015). Bootstrap support was assessed using an ultrafast bootstrap (BP) with 1000 replicates (Hoang et al. Citation2018). Overall, the relationships between the three lineages (Echiura, Errantia and Sedentaria) followed a well-established annelid phylogeny (Weigert et al. Citation2016). As the tree indicated, M. japonicus was closely related to Chaetopterus variopedatus and Phyllochaetopterus sp., all of which belong to Chaetopteridae, which is the sister taxon to other Sedentaria taxa.
Figure 1. The Maximum-likelihood (ML) phylogenetic tree for M. japonicus and the other Polychaeta species based on the concatenated nucleotide sequences of 13 protein-coding genes, and M. japonicus is placed with Sedentaria. Bootstrap support values are indicated at each node.
Author contributions
Mei Yang analyzed the data, prepared the figures and wrote drafts of the paper. Weina Wang collected the samples and analyzed the data. Xinzheng Li conceived, designed and supervised the work. Jixing Sui supervised the work and revised it critically for intellectual content. All authors have agreed to the published version of the manuscript and to be accountable for all aspects of the work.
Disclosure statement
The authors report no conflict of interest.
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov under the accession no. MZ921947. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA801441, SRR17798510, and SAMN25349843, respectively.
Additional information
Funding
References
- Barr CM, Maurine N, Taylor DR. 2005. Inheritance and recombination of mitochondrial genomes in plants, fungi and animals. New Phytol. 168(1):838–50.
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319.
- Boore JL. 1999. Animal mitochondrial genomes. Nucleic Acids Res. 27(8):1767–1780.
- Fujiwara T. 1934. On a new chaetopterid, Mesochaetopterus japonicus sp. nov. J Sci Hiroshima Univ Ser B Div I Zool. 3:1–14.
- Gissi C, Iannelli F, Pesole G. 2008. Evolution of the mitochondrial genome of metazoa as exemplified by comparison of congeneric species. Heredity. 101(4):301–320.
- Hao W, Richardson AO, Zheng Y, Palmer JD. 2010. Gorgeous mosaic of mitochondrial genes created by horizontal transfer and gene conversion. Proc Natl Acad Sci U S A. 107(50):21576–21581.
- Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS. 2018. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol. 35(2):518–522.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Lee H, Chen WJ, Puillandre N, Aznar-Cormano L, Tsai MH, Samadi S. 2019. Incorporation of deep-sea and small-sized species provides new insights into gastropods phylogeny. Mol Phylogenet Evol. 135:136–147.
- Luo RB, Liu BH, Xie YL, Li ZY, Huang WH, Yuan JY, He GZ, Chen YX, Pan Q, Liu YJ, et al. 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 1(1):18–23.
- Moore JM, Nishi E, Rouse GW. 2017. Phylogenetic analyses of Chaetopteridae (Annelida). Zool Scr. 46(5):596–610.
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Nishi E. 1999. Redescription of Mesochaetopterus selangolus (Polychaeta: Chaetopteridae), based on type specimens and recently collected material from Morib Beach, Malaysia. Pac Sci. 53(1):24–36.
- Nishi E, Hsieh HL. 2009. Chaetopterid polychaetes from Taiwan and Okinawa Island (Japan), with descriptions of two new species. Zool Stud. 48(3):370–379.
- Shen Y, Kou Q, Zhong Z, Li X, He L, He S, Gan X. 2017. The first complete mitogenome of the South China deep-sea giant isopod Bathynomus sp. (Crustacea: Isopoda: Cirolanidae) allows insights into the early mitogenomic evolution of isopods. Ecol Evol. 7(6):1869–1881.
- Weigert A, Golombek A, Gerth M, Schwarz F, Struck TH, Bleidorn C. 2016. Evolution of mitochondrial gene order in Annelida. Mol Phylogenet Evol. 94(Pt A):196–206.
- Yang D, Sun R. 1988. Polychaetous annelids commonly seen from the Chinese waters (in Chinese). Beijing: China Agriculture Press, p. 352.