
Abstract
Rubus peltatus Maxim. (Bull. Acad. Imp. 1871) is a wild species endemic to East and Southeast China. However, genetic resources were unavailable for this species. It holds great potential for domestication or other breeding purposes with the extraordinary large yellow fruits. The complete chloroplast genome sequence of R. peltatus, assembled with Illumina Hiseq X Ten platform sequencing data, was reported. The chloroplast genome was 155,582 bp in length. The large single-copy (LSC) and small single-copy (SSC) of 85,329 bp and 18,779 bp were separated by two inverted repeats (IRs) of 25,737 bp. The chloroplast genome of R. peltatus contains 130 genes, including eight transfer RNA genes, 36 ribosomal RNA genes, and 86 protein-coding genes. Phylogenetic analysis supports R. peltatus has a close relationship with the R. cochinchinensis and R. takesimensis.
Rubus Linnaeus is a conspicuous genus in the family of diverse raspberries and blackberries, which are popular fruits around the world with the low calories and excellent sources of fiber, vitamin C, and antioxidant compounds. Being the diversity center for the Rubus genus, China has about 204 species, accounting for 97% of Asian species (Lu Citation1983). However, few wild Rubus species has been domesticated or utilized so far. The species Rubus peltatus Maxim. is endemic to East and Southeast China, and is promising for domestication with extraordinary large yellow fruits. However, genetic resources were unavailable for this species. In this study, we reported and characterized the chloroplast genomes of R. peltatus for further study and utilization of this species.
Plant samples of R. peltatus were collected in the field from Guizhou province, China (27°54′N, 108°34′E, 1018 m above sea level). The voucher specimen was deposited at the Herbarium of Sun Yat-sen University (SYS) with accession number SYS00001512 (Weicheng Huang, [email protected]). Total genomic DNA was extracted from silica gel dried leaves using a modified CTAB protocol (Doyle Citation1991). After agarose gel electrophoresis for inspection of integrity and quantification, the total DNA was sent to Berry Genomics Bio-Technique Co. Ltd. (Beijing, China) for sequencing service. A fragment library with insertion size of 500 bp was prepared and sequenced using paired-end reads 150 bp in length on the Illumina Hiseq X Ten platform (Illumina, Inc., San Diego, CA). Using fastp v0.20.1 (Chen et al. Citation2018), raw-reads were subject to adapter cutting without further trimming or filtering as recommended by the de novo assembler NOVOPlasty (Dierckxsens et al. Citation2017). Next, setting the whole chloroplast genome sequences of Rubus leucanthus (MK105853.1) as seed, we assembled the R. peltatus chloroplast genome using an extension algorithm implemented in NOVOPlasty with default settings. The assembled cp genome was annotated using GeSeq (Tillich et al. Citation2017), and the annotation was corrected using Geneious v.9.0.2 (Kearse et al. Citation2012). The complete genome sequence and annotations of R. peltatus were submitted to GenBank under accession number NC_056937.1 (Wei Guo, [email protected]).
The complete cp genome of R. peltatus was 155,582 bp in length. The typical structure consists of a large single-copy (LSC) of 85,329 bp and a small single-copy (SSC) of 18,779 bp, separated by two inverted repeats (IRs) (25,737 bp). The chloroplast genome circle was joined end-to-end in the order ‘LSC-IRA-SSC-IRB’. The overall GC content of cp genome was 36.91%. There were 130 predicted genes, 86 protein-encoding genes, 36 tRNA genes, and eight rRNA genes.
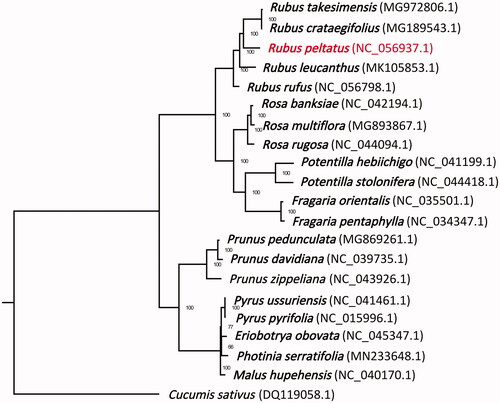
To confirm the phylogeny position of R. peltatus in Rosaceae, 19 representative species within family Rosaceae were obtained from GenBank database (https://www.ncbi.nlm.nih.gov/). Cucumis sativus (DQ119058.1) was specified as one outgroup taxon. The sequences were aligned using MAFFT v.7.313 (Katoh and Standley Citation2013). A maximum-likelihood tree was constructed in the package RAxML v.8.2.11 (Stamatakis Citation2014) using Cucumis sativus as outgroup with bootstrap replicates of 100 (). Phylogenetic analysis demonstrated that R. peltatus was placed in the Rubeae tribe with a closer relationship with R. cochinchinensis and R. takesimensis, than that with R. leucanthus or R. rufus.
Figure 1. Maximum-likelihood tree of Rosaceae based on complete chloroplast genome, with Cucumis sativus as outgroup. The Rubus peltatus Maxim. is marked in red and bootstrap support values are shown next to the nodes.
Ethics statement
The samples used in our study are common species in the field, and we only sampled the leaves. There’s no need for permission.
Author contributions
LW conceived and initiated this research. FQ and WCH performed data analysis. WCH, WKD, and WG collected the samples. FQ wrote the manuscript.
Acknowledgements
We are grateful to Prof. Wei Wu and Dr. Can Lai for critical reading of English of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov/ under the accession no. NC_056937.1. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA747400, SRR12424495, and SAMN15763317, respectively.
Additional information
Funding
References
- Chen S, Zhou Y, Chen Y, Gu J. 2018. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 34(17):i884–i890.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Doyle JJ. 1991. DNA protocols for plants—CTAB total DNA isolation. In: Hewitt GM, Johnston A, editors. Molecular techniques in taxonomy. Berlin: Springer; p. 283–293.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Lu LD. 1983. A study on the genus Rubus of China. J Syst Evol. 21:13–25.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq- versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45(W1):W6–W11.