
Abstract
Vincetoxicum junzifengense B.J. Ye and S.P. Chen 2022 is a newly described species which belongs to the genus Vincetoxicum in the family Apocynaceae. The complete plastid genome of Vincetoxicum junzifengense B.J. Ye and S.P. Chen 2022 was determined and analyzed in this study. The total chloroplast genome was 159,666 bp in length, consisting of a large single-copy region of 90,565 bp, a small single-copy region of 19,691 bp, and two inverted repeat regions of 24,705 bp. The genome contained 131 genes, including 85 protein-coding genes, 37 transfer RNA genes, and eight ribosomal RNA genes. Phylogenetic analysis indicated that V. junzifengense is sister to V. versicolor.
Vincetoxicum, which consists of approximately 200 species, is one of the most species-rich genera of Apocynaceae (Shah et al. Citation2021). They are distributed in Asia, Africa, Arabia, Australia, and Eurasia (Douglass et al. Citation2009) and China is one of the most diverse centers, with 71 species of the Vincetoxicum genus distributed in China (Jiang et al. Citation2018). Vincetoxicum junzifengense B.J. Ye and S.P. Chen Citation2022 is a newly described species and characterized by its fascicled and slender roots, narrowly lanceolate leaf blade, raceme-like inflorescences, amaranth corolla with triangular lobes, cupular corona with triangular and dentate lobes, pentagon and slightly elevated stigma, two ovoid pollinia per pollinarium, single lanceolate and glabrous follicles. This species was only found at one site in the Junzifeng National Nature Reserve, West Fujian, China, and grows in the subtropical evergreen broad-leaved forest, moist and loose soil. From morphological aspects, V. junzifengense is similar to V. versicolor and V. stauntonii and our phylogenetic analysis supported that V. junzifengense is a sister to V. versicolor (Ye et al. Citation2022). In this study, the complete plastid genome of this species was assembled, annotated and analyzed.
The samples were collected from Mingxi district, Fujian, China (25°35′N, 116°55′E) and the voucher specimen deposited at Herbarium of Fujian Agriculture and Forestry University (http://lxy.fafu.edu.cn/, contact person: Chen Shipin; email: [email protected]) under the voucher code Baojian Ye WSD9808. Total genomic DNA was extracted from fresh leaves using a modified CTAB method of Doyle (Doyle Citation1987) and sequenced by the BGISEQ-500 platform. The GetOrganelle v1.7.5 (Jin et al. Citation2020) was used to filter the raw sequence reads to get the high-quality plastid like reads. And then the target-associated reads produced by the former step to get the final FASTA files were assembled using SPAdes within the same pipeline (Bankevich et al. Citation2012). With the chloroplast genome of Vincetoxicum versicolor (GenBank accession NC_052877) as the reference sequences, the assembled plastid genome was annotated using the Geneious R11.15 (Kearse et al. Citation2012).
The total plastid genome of V. junzifengense (GenBank accession OM995819) was 159,666 bp in length, consisting of a large single-copy (LSC) region of 90,565 bp, a small single-copy (SSC) region of 19,691 bp, and two inverted repeat regions (IRA and IRB) of 24,705 bp. The GC content of the chloroplast genome was 37.80%, while the corresponding values of the LSC, SSC, and IR regions are 56.72%, 12.33%, and 30.95%, respectively. The complete chloroplast genome contains 131 genes, including 85 protein-coding genes, 37 transfer RNA (tRNA) genes, and eight ribosomal RNA (rRNA) genes.
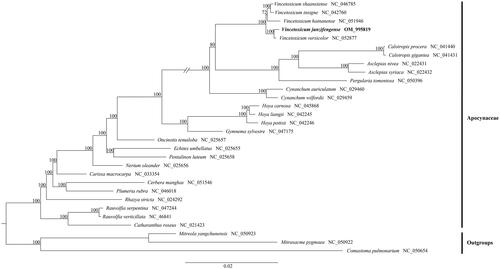
To detect the phylogenetic relationship of V. junzifengense with other Apocynaceae members, additional 27 representative species of Apocynaceae were downloaded from NCBI, with two species of Loganiaceae and one species of Gentianaceae as outgroups. Data matrices were aligned by MAFFT v7 (Katoh and Standley Citation2013) plugin in the software Phylosuite v1.2.2 (Zhang et al. Citation2020). After nucleotide sequence alignment, the phylogenetic analysis was performed based on the complete plastid genomes using the maximum likelihood (ML), which was conducted on the website CIPRES Science Gateway with RAxML-HPC2 on XSEDE 8.2.10 (Miller et al. Citation2010), and the phylogenetic tree constructed by RAxML (Stamatakis Citation2014) with 1,000 bootstrap replicates (Minh et al. Citation2013; Chernomor et al. Citation2016). Our molecular analysis indicates that V. junzifengense was sister to V. versicolor with strongly support (BP = 100) ().
Figure 1. The maximum-likelihood (ML) tree based on the 27 representative plastid genome sequences of Apocynaceae and two outgroup species of Loganiaceae and one outgroup species of Gentianaceae. The bootstrap value near each node.
Ethical approval
The sample collection was authorized by Xue-Fen Cao (Junzifeng National Natural Reserve).
Author contributions
Bao-jian Ye conceived this study and drafted the manuscript; Xia-bing Shen assisted in editing and revised the manuscript for intellectual content; Yu-wei Wu performed the data analysis; Xue-Fen Cao conducted sample collection from wild fields; Xing-wen Zhou designed this study, administrated the project and provided financial support; all authors contributed to the final approval of the version to be published and all agree to be accountable for all aspects of the work.
Disclosure statement
The authors declare that there are no conflicts of interest regarding the publication of this article. The authors alone are responsible for the content and writing of the paper.
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at (https://www.ncbi.nlm.nih.gov/) under the accession no. OM995819.1. The associated BioProject, Bio Sample and SRA, numbers are PRJNA845946, SAMN28865336, and SRR19543586 respectively.
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Chernomor O, von Haeseler A, Minh BQ. 2016. Terrace aware data structure for phylogenomic inference from supermatrices. Syst Biol. 65(6):997–1008.
- Douglass CH, Weston LA, DiTommaso A. 2009. Black and pale swallow-wort (Vincetoxicum nigrum and V. rossicum): the biology and ecology of two perennial, exotic and invasive vines. Management of Invasive Weeds. 5:261–278.
- Doyle J. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Jiang LQ, Li YY, Zhu XX, Wang YH, Peng H. 2018. Vincetoxicum xinpingense (Asclepiadeae, Asclepiadoideae, Apocynaceae), a new species from Yunnan Province, China. Phytotaxa. 361(1):56–64.
- Jin JJ, Yu WB, Yang JB, Song Y, DePamphilis CW, Yi TS, Li DZ. 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):1–31.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Miller MA, Pfeiffer W, Schwartz T. 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Gatew Comput Environ Work (GCE). 14:1–8.
- Minh BQ, Nguyen MAT, von Haeseler A. 2013. Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol. 30(5):1188–1195.
- Shah SA, Sultan A, Wen J, Ullah Z, Nisa SU, Ren Z, Alam MM, Iqbal J, Mumtaz AS. 2021. Taxonomy of Vincetoxicum s. str. (Asclepiadoideae, Apocynaceae) from southern Asia including three new species and resurrected names. PhytoKeys. 179:35–73.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Ye BJ, Tu XD, Zhou CY, Liu B, Cao XF, Chen SP. 2022. Vincetoxicum junzifengense (Apocynaceae), a new species from Fujian, China: evidence from morphological and molecular analyses. Phytotaxa. 539(2):203–209.
- Zhang D, Gao F, Jakovlic I, Zou H, Zhang J, Li WX, Wang GT. 2020. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour. 20(1):348–355.