
Abstract
Pellionia scabra Benth. 1861 (Urticaceae) is distributed in east and southeast Asian countries, including China, Vietnam, and Japan, and has important applications in construction, medicine, and the food industry. We sequenced the genome using Illumina DNA sequencing technology. The genome was 153,220 bp long. Annotation of the genome showed that it encoded 130 genes, including 85 protein-coding genes, 37 tRNA genes, and eight rRNA genes. In addition, 15 of the genes contained a single intron and two contained two introns. Furthermore, rps12 consists of 3 exons that are expected to be trans-spliced together. Subsequent phylogenetic analysis revealed that P. scabra is closely related to the Elatostema species (i.e. E. stewardii [MZ292972], E. dissectum [MK227819], and E. laevissimum var. laevissimum [MN189961]).
Pellionia scabra Benth. 1861 (Urticaceae) is distributed in east and southeast Asian countries, including China, Vietnam, and Japan. The species is popular for its use in interior decoration and as a source of traditional Chinese medicine. In addition, P. scabra is a popular wild vegetable (Wang et al. Citation2013). The aim of the present study was to improve the current understanding of the species’ characteristics and functions by sequencing its complete chloroplast (cp) genome and by performing a phylogenetic analysis of 77 common protein-coding genes from the cp genomes of P. scabra and 28 other species.
Leaves of P. scabra were collected from Guizhou Botanical Garden (N 26°37′20″, E 106°43′29″) and stored samples (accession no. MCC20210902YX) from the Laboratory of the College of Agriculture, Guizhou University (Guiyang, China; contact person: Xuelian Yang, email: [email protected]). The study was approved by Guizhou University and performed in accordance with the national Wild Plant Protective Regulations.
Total genomic DNA was extracted from 150 mg fresh leaves using the cetyltrimethylammonium bromide method (Doyle and Doyle Citation1987). An aliquot of purified DNA (0.5 μg) was then fragmented to construct a short-insert (350 bp) library using the Nextera XT DNA library preparation kit (Illumina, San Diego, CA). The library was sequenced using the Illumina NovaSeq 6000 platform, and the raw data were edited using the NGS QC Tool Kit v2.3.3 (Patel and Jain Citation2012). High-quality sequence data (3912 Mb) were then selected for the de novo assembly of the complete cp genome using the assembler SPAdes v3.11.0 (Bankevich et al. Citation2012). Finally, the complete cp genome was annotated using PGA (Qu et al. Citation2019) with the cp genome of Elatostema stewardii as a reference.
The complete cp genome of P. scabra (153,220 bp) exhibited a typical quadripartite structure, including a large single-copy (LSC) region (84,480 bp), small single-copy (SSC) region (17,568 bp), and pair of inverted repeat (IR) regions (IRa and IRb, each 25,586 bp). The cp genome’s total GC content was 36.4%, and the genome contained 130 genes, including 85 protein-coding genes, 37 tRNA genes, and eight rRNA genes. In addition, 15 of the genes (trnK-UUU, rps16, trnG-UCC, atpF, rpoC1, trnL-UAA, trnV-UAC, petB, petD, rpl16, rpl2, ndhB, trnI-GAU, trnA-UGC, and ndhA) contained a single intron and two (ycf3 and clpP) contained two introns. Furthermore, rps12 consists of three exons that are expected to be trans-spliced together.
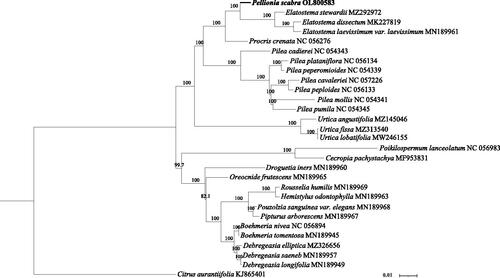
To analyze the P. scabra cp genome, the complete cp genomes of 28 other plant species, including 27 other members (16 genera) of Urticaceae and one outgroup taxon (Citrus aurantifolia, Rutaceae; KJ865401), were obtained from NCBI. The sequences of 77 common protein-coding genes from P. scabra cp genome and the other 28 other complete cp genomes were aligned using MAFFT version 7.037 (Katoh and Standley Citation2013) with the FFT-NS-2 strategy. A phylogenetic tree of the 29 cp genomes was then constructed using RAxML version 8.2.9 (Stamatakis Citation2014), based on the maximum-likelihood method with 1000 bootstrap replications and the TVM + F+I + G4 model, which was selected using ModelFinder version 1.6 (Kalyaanamoorthy et al. Citation2017).
Phylogenetic analysis indicated that P. scabra is closely related to the Elatostema species (i.e. E. stewardii [MZ292972], E. dissectum [MK227819], and E. laevissimum var. laevissimum [MN189961]; ). In addition, Pellionia and Elatostema were more closely related to the genera Procris, Pilea, and Urtica than to the other genera, and the genera Oreocnide, Rousselia, Hemistylus, Pouzolzia, Pipturus, Boehmeria, Debregeasia, and Droguetia formed a distinct clade.
Figure 1. Maximum-likelihood tree of protein-coding gene sequences from the complete chloroplast sequences of Pellionia scabra Benth.1861 (Urticaceae) and 28 other plant species. The analysis was performed using 77 homologous protein-coding genes. Node values indicate bootstrap support (1000 replicates).
The results of this study provide information about the complete cp genome of P. scabra, thereby facilitating further research on the species’ clinical and commercial applicability.
Author contributions
Xue-Lian Yang conceived and designed the study; Xue-Lian Yang, Xia Wang, Yong-Fei Wu, Xiao-Jing Hu, and Li Yan collected the samples, performed the experiments, and analyzed the data; Xia Wang and Xiao-Jing Hu drafted and revised the manuscript. All authors approved the final manuscript and agreed to be accountable for all aspects of the work.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank (https://www.ncbi.nlm.nih.gov/) under accession number OL800583. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA787431, SRR17177970, and SAMN23802974, respectively.
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kalyaanamoorthy S, Minh BQ, Wong TK, von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589.
- Patel RK, Jain M. 2012. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLOS One. 7(2):e30619.
- Qu XJ, Moore MJ, Li DZ, Yi TS. 2019. PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods. 15(1):1–12.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Wang L, Liao W, Qiu W. 2013. Nutritious components of the wild vegetable Pellionia scabra from South China. Acta Scientiarum Naturalium Universitatis Sunyatseni. 000(006):119–123.