
Abstract
The complete mitochondrial genome of Nadezhdiella cantori (Hope, 1843) is 16,049 bp in length, containing 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNAs), two ribosomal RNA genes (rRNAs), and an A-T rich region (control region). The gene order is conserved and identical to most other previously sequenced Cerambycidae. Phylogenetic analysis showed that the newly sequenced N. cantori was in a well-supported clade tribe Cerambycini, subfamily Cerambycinae. These results support the currently accepted taxonomy and provide a better understanding of the phylogenetic analysis of the Cerambycidae.
Introduction
Family Cerambycidae, known as longhorn beetles or longicorn beetles, contains over 30,000 species worldwide. There are more than 12,000 species described in subfamily Cerambycinae, the second largest subfamily. The phylogenetic relationship of Cerambycinae remains unclear (Lee and Lee Citation2020). A recent study suggested that mitochondrial genomics are useful for revealing phylogenetic relationships (Nie et al. Citation2021). According to the data from GenBank, the complete mitogenomes of 27 species of Cerambycinae have been reported so far. Nadezhdiella cantori (Hope, 1843), belonging to subfamily Cerambycinae, is an important citrus pest that feeds on the trunks and main branches of citrus trees (Wang and Zeng Citation2004) and is widely distributed in China, Laos, Thailand, and Vietnam. However, the complete mitochondrial genome of N. cantori has not been reported. In the present study, the characterization of the mitogenome of N. cantori is reported.
Materials and methods
Specimens of N. cantori were collected from Jinyun Mountain National Nature Reserve, Chongqing, China (106°22′E, 29°49′N) on 6 June 2021 and were deposited at the Entomological Collection of Southwest University, Chongqing, China (voucher number SWU-01021601. Zhu Li, email: [email protected]). The sample was preserved in 100% ethanol at −20 °C ().
Figure 1. Adult habitus of Nadezhdiella cantori (Hope, 1843).

Genomic DNA was extracted from an adult’s muscle tissue of the prothorax or leg by the CTAB method (Reineke et al. Citation1998). Genomic DNA was subjected to pin-end (PE) sequencing based on the Illumina NovaSeq sequencing platform using next-generation sequencing technology at Personalbio (Shanghai, China). The data obtained by sequencing were used to assemble the mitochondrial genome using GetOrganelles (Jin et al. Citation2020). The assembled mitochondrial genome was annotated by using MITOS Web Server (Bernt et al. Citation2013) and Geneious (Kearse et al. Citation2012). Bayesian’s inference was performed using MrBayes v.3.2 (Ronquist et al. Citation2012) with the best model of the gene fragment GTR + F+I + G4 (atp6, cox1, cox2, cox3, cytb, nad1, nad2, nad3, nad4, nad4L, and nad5), and HKY + F+I + G4 (atp8 and nad6) was determined by ModelFinder (Kalyaanamoorthy et al. Citation2017).
Results
The complete mitogenome of N. cantori (GenBank accession number NC_061180) is 16,049 bp in length, containing the entire set of 37 genes usually present in most insect mtDNAs (13 protein-coding genes (PCGs), 22 transfer RNA genes, and two ribosomal RNAs), and a putative control region (). Fourteen genes were transcribed on the minority strand (N-strand), whereas the others were oriented on the majority strand (J-strand). The overall base composition of the N. cantori mitogenome was A (40.1%), T (31.9%), C (17.2%), and G (10.8%), with a high A + T bias of 72.00%.
Figure 2. Genome map of the mitogenome of N. cantori.
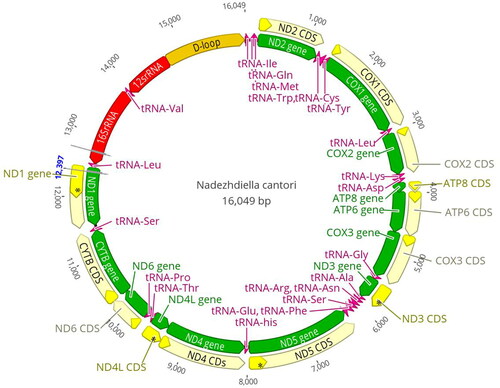
A total of 31 bp intergenic spacer were dispersed in six locations, ranging from 1 to 22 bp in length. The longest interval between trnS2 and nad1 was 22 bp. Gene overlaps were found at 14 gene junctions and involved a total of 47 bp, the longest 8 bp overlap located between trnY and cox1. The length of 22 tRNAs varied between 64 bp (trnT, trnH, trnF, trnE, trnR, and trnG) and 71 bp (trnK), comprising a total of 1456 bp. The length of rrnL is 1289 bp with an A + T content of 76%, and rrnS is 804 bp with an A + T content of 70.5%. The 1356 bp control region is located between rrnS and trnI and has a remarkably high A + T content (81.8%).
Twelve PCGs begin with ATN start codons, excluding nad1, which begins with TTG: ATG for cox3, nad4, nad4l, atp6, and cytb; ATT for cox1, nad2, and nad6; ATA for nad3 and nad5; ATC for cox2 and atp8. The PCGs terminate with a TAN (TAA or TAG) stop codon, whereas five PCGs (cox2, cox3, nad3, nad4, and nad5) terminate with an incomplete stop codon (T–). For the seven other PCGs, TAAs (nad2, nad6, atp8, atp6, nad4L, and cox1) or TAGs (cytb and nad1) were used.
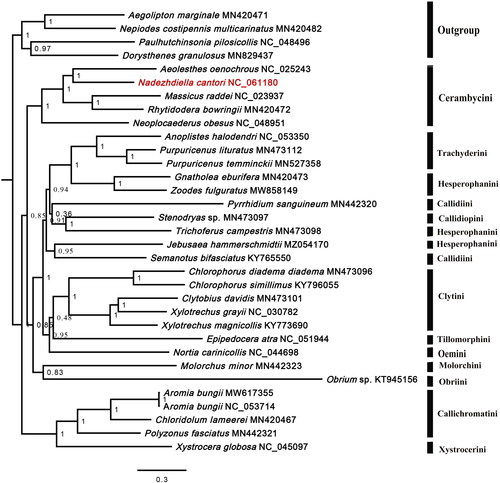
We reconstructed the phylogenetic relationships of 28 Cerambycinae species based on 13 PCGs. The Bayesian tree () showed that Cerambycinae is monophyletic. Most tribes are monophyletic, whereas Hesperophanini and Callidiini are not. N. cantori formed a sister to Aeolesthes oenochrous with high value support.
Figure 3. Bayesian tree of 28 species of Cerambycinae, including Nadezhdiella cantori (in this study, NC_061180) and 4 outgroups based on the sequence of 13 mitochondrial protein-coding genes. Numbers on each node are posterior probabilities.
Discussion and conclusions
This study determined the gene sequence of N. cantori and analyzed the main characteristics of its mitochondrial genome. The mitochondrial genome is conserved in terms of gene composition, location, and direction, which are the same as other previously sequenced mitochondrial genomes of Cerambycidae (Yang et al. Citation2019; Li et al. Citation2021).
Phylogenetic analysis showed that Cerambycinae is monophyletic. Hesperophanini and Callidiini are not, which is consistent with the previous studies (Lee and Lee Citation2020; Nie et al. Citation2021). In its present phylogenetic position, N. cantori is clustered within Cerambycini species, consistent with its classification based on morphological data. The mitogenome of N. cantori will help with molecular identification and population genetic studies of the species. It also provides a basis for establishing the phylogenetic relationships of Cerambycinae.
Ethics statement
This study did not need ethical approval or permission to collect the sample. The sequenced species is common in China and is not included in the ‘List of Protected Animals in China’. All of the research meets ethical guidelines and adheres to the legal requirements of the country the study was conducted in.
Author contributions
L.Z. collected the specimen, conceptualized and designed the idea; T.H. analyzed and interpreted the data; T.H. was responsible for the drafting of the paper; L.Z. was responsible for the revising it and for the final approval; all authors agreed to be accountable for all aspects of the work.
Disclosure statement
The authors declare that there is no potential conflict of interest.
Data availability statement
The mitogenome sequence data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov/ under the accession no. NC_061180. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA835288, SRR19143081, and SAMN28100428, respectively.
Additional information
Funding
References
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319.
- Jin JJ, Yu WB, Yang JB, Song Y, dePamphilis CW, Yi TS, Li DZ. 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):241.
- Kalyaanamoorthy S, Minh BQ, Wong T, Haeseler A, Jermiin L. 2017. Model Finder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Lee S, Lee S. 2020. Multigene phylogeny uncovers oviposition-related evolutionary history of Cerambycinae (Coleoptera: Cerambycidae). Mol Phylogenet Evol. 145:106707.
- Li MR, Song X, Du Y. 2021. Mitochondrial genome of Aromia bungii (Coleoptera: Chrysomeloidea: Cerambycidae) and phylogenetic analysis. Mitochondrial DNA Part B. 6(1):71–72.
- Nie R, Vogler AP, Yang XK, Lin MY. 2021. Higher-level phylogeny of longhorn beetles (Coleoptera: Chrysomeloidea) inferred from mitochondrial genomes. Syst Entomol. 46(1):56–70.
- Reineke A, Karlovsky P, Zebitz CPW. 1998. Preparation and purification of DNA from insects for AFLP analysis. Insect Mol Biol. 7(1):95–99.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61(3):539–542.
- Wang Q, Zeng W. 2004. Sexual selection and male aggression of Nadezhdiella cantori (Hope) (Coleoptera: Cerambycidae: Cerambycinae) in relation to body size. Environ Entomol. 33(3):657–661.
- Yang WJ, Yan SY, Xu KK, Li C. 2019. Characterization of the complete mitochondrial genome of Purpuricenus temminckii (Coleoptera: Cerambycidae). Mitochondrial DNA Part B. 4(2):3994–3995.