
Abstract
Here, we sequenced and annotated the complete mitochondrial genome for the terebellid polychaete, Neoamphitrite affinis (Malmgren 1866). The complete mitogenome of N. affinis is 15,823 bp, with 33.4% A, 18.2% C, 11.5% G, and 37.0% T. The circular N. affinis mitochondrial genome comprises 13 protein-coding genes (PCGs), 23 transfer RNA (tRNA) genes including 2 methionine tRNA genes, 2 ribosomal RNA (rRNA) genes, and a non-coding region. Phylogenomic analysis based on 26 in-group taxa belonging to the two main clades, Sedentaria and Errantia, is congruent with published phylogenetic relationship for annelids, which N. affinis was grouped with Pista cristata (Terebellida; Terebellidae). This mitogenome resource will be useful for future phylogenetic studies of families belonging to Sedentaria.
The phylum Annelida has two major phylogenetic groups, Pleistoannelida, and six early-branching lineages such as Amphinomida, Chaetopteridae, Lobatocerebrum, Magelonidae, Oweniidae, and Sipuncula (Weigert and Bleidorn Citation2016), although the phylum is still complex and detailed phylogeny remains debated due to notably derived morphologies, remarkable ecological diversity, and the lack of genomic information (Struck et al. Citation2011; Weigert et al. Citation2014; Weigert and Bleidorn Citation2016; Struck Citation2017). Two main monophyletic annelid backbones, Sedentaria and Errantia have well-established in Pleistoannelida (Kvist and Siddall Citation2013; Weigert et al. Citation2014; Weigert and Bleidorn Citation2016; Struck Citation2017), but detailed phylogenetic relationship on families of Sedentaria remained controversial. Members in the sedentarian family Terebellidae are known as spaghetti worms and are ubiquitous tubicolous benthic annelids with morphological and ecological diversity (Stiller et al. Citation2020). Recently, the monophyly of major taxa including Terebellidae was restored based on transcriptome information and morphological characters (Stiller et al. Citation2020), whereas there are only two complete mitogenome pieces of information belonging to the family Terebellidae available: Pista cristata and Thelepus plagiostoma (Nam et al. Citation2021).
The genus Neoamphitrite Hessle Citation1917, distinguished from the genus Amphitrite Müller, 1771 (Hessle Citation1917) by the morphometric characteristics of branchiae and nephridia (Hessle Citation1917; Hutchings and Glasby Citation1988), inhabits soft sediments from intertidal to the shallow offshore reef with a wide range of distribution. In this study, a specimen of N. affinis was sampled from the Beaufort Sea (69°52′N, 139°03′W) in 2017 using a remotely operated underwater vehicle (ROV) belonging to the Monterey Bay Aquarium Research Institute (MBARI). All animal handling and experimental procedures were approved by the Animal Welfare Ethical Committee and the Animal Experimental Ethics Committee of the Incheon National University (Incheon, South Korea). The specimen and extracted DNA sample were deposited in the Research Institute of Basic Sciences of Incheon National University (Species ID: Annelid-06; Specimen ID: KOPRI-Benthos-06; https://www.inu.ac.kr/user/indexMain.do?siteId=ribs; Dr. Sang-Eun Nam; [email protected]). Total genomic DNA was isolated from the muscle tissue of a single individual using a DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Next-generation sequencing was performed to obtain a circular mitogenome with the HiSeq platform (150 bp; HiSeq X ten; Illumina, San Diego, CA, USA) using previously described protocols (Nam et al. Citation2021). After the quality check, 26,605,410 filtered reads (3,999,082,033 bp) were obtained from 33,789,776 raw reads (5,102,256,176 bp). Thereafter, de novo assembly was conducted with various k-mers using SPAdes (Bankevich et al. Citation2012) to obtain a circular contig of the N. affinis mitogenome. The resulting contig consensus sequence was annotated using MITOS2 (Bernt et al. Citation2013) and tRNAscan-SE 2.0 (Lowe and Eddy Citation1997). BLAST searches (http://blast.ncbi.nlm.nih.gov) and multiple alignments with terebellids’ mitogenomes confirmed the identity of these genes.
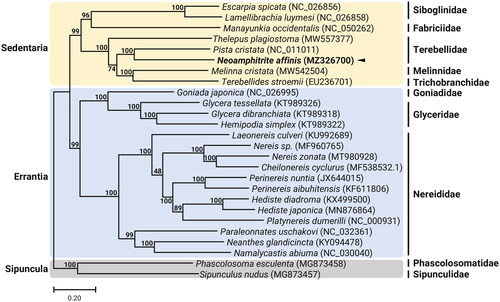
The complete mitogenome of N. affinis is 15,823 bp in length (GenBank accession no. MZ326700) with the typical composition, consisting of 13 PCGs, 23 tRNAs, 2 rRNAs, and a large intergenic region presumed to be the control region. However, in addition to the 22 tRNAs, we recover an additional methionine tRNA (trnM), which is located next to the first trnM gene in tandem. This unique addition of the trnM gene in tandem was previously observed in the mitogenomes of Pista cristata (Terebellidae) and Terebellides stroemi (Trichobranchidae) (Zhong et al. Citation2008), whereas this phenomenon was not conserved in the family Terebellidae, as Thelepus plagiostoma mitogenome contained single trnM gene (Nam et al. Citation2021). Duplication of the trnM has also been detected in the mitogenomes of other polychaetes such as Eurythoe complanata (Amphinomidae) and Typosyllis antoni (Syllidae) (Weigert et al. Citation2016). The nucleotide composition of N. affinis mitogenome was highly biased toward A + T nucleotides (70.3%), with percentages of A, T, C, and G being 33.4%, 37.0%, 18.2%, and 11.5%, respectively. The COI sequence showed the highest similarity (99.9%) to the COI sequence of N. affinis previously registered in GenBank (657 bp; GenBank accession no. MT167006). We constructed the phylogenetic topology of 26 members belonging to Sedentaria and Errantia using the concatenated set of 13 PCG sequences, with two species as an outgroup (). Neoamphitrite affinis is closely related to P. cristata (Terebellidae) within Terebellidae.
Figure 1. Maximum-likelihood (ML) phylogeny of 8 published mitogenomes from Sedentaria including T. plagiostoma and 16 registered mitogenomes of Errantia species, and two Sipuncula species as an outgroup based on the concatenated nucleotide sequences of protein-coding genes (PCGs). The phylogenetic analysis was performed using the maximum-likelihood method, GTR + G+I model with a bootstrap of 1000 replicates. Numbers on the branches indicate ML bootstrap percentages. DDBJ/EMBL/Genbank accession numbers for published sequences are incorporated. The black triangle means the polychaete analyzed in this study.
Ethical approval
This study did not involve endangered or protected species, and the sample was collected from the Beaufort Sea (69°52′N, 139°03′W) and transferred in accordance with guidelines and in compliance with research legislation. All the experiments using the fish were approved by the animal care and use committee of Incheon National University (Incheon, South Korea).
Authors’ contributions
S.-E. Nam: conceptualization, methodology, software, writing; S. Lee: methodology, software, data curation; Y. Lee: methodology, software; J.-S. Rhee: conceptualization, supervision, reviewing, and editing.
Acknowledgments
The authors wish to thank Dr. Tae-Yoon S. Park and Mr. Ji-Hoon Kihm (Korea Polar Research Institute) for their help in fieldwork and sampling.
Disclosure statement
The authors report no conflicts of interest and are solely responsible for the content and writing of this manuscript. All the authors agreed to be accountable for all aspects of the work.
Data availability statement
BioProject, BioSample, and SRA accession numbers are https://www.ncbi.nlm.ni h.gov/bioproject/PRJNA739841, https://www.ncbi.nlm.nih.gov/biosample/SAMN19803891, and https://www.ncbi.nlm.nih.gov/sra/?term=SRR15041508, respectively. The data that support the findings of this study are openly available in the National Center for Biotechnology Information (NCBI) at https://www.ncbi.nlm.nih.gov, with an accession number MZ326700.
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Bernt A, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319.
- Hessle C. 1917. Zur Kenntnis der terebellomorphen Polychaeten. Zoologiska Bidrag Från Uppsala. 5:39–258.
- Hutchings PA, Glasby CJ. 1988. The Amphitritinae (Polychaeta: Terebellidae) from Australia. Rec Aust Mus. 40(1):1–60.
- Kvist S, Siddall ME. 2013. Phylogenomics of Annelida revisited: a cladistics approach using genome-wide expressed sequence tag data mining and examining the effects of missing data. Cladistics. 29(4):435–448.
- Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25(5):955–964.
- Nam S-E, Park HS, Kim SA, Kim B-M, Rhee J-S. 2021. The complete mitochondrial genome of the terebellid polychaete Thelepus plagiostoma (Terebellida; Terebellidae). Mitochondrial DNA Part B. 6(11):3114–3116.
- Stiller J, Tilic E, Rousset V, Pleijel F, Rouse GW. 2020. Spaghetti to a tree: a robust phylogeny for Terebelliformia (Annelida) based on transcriptomes, molecular and morphological data. Biology. 9(4):73.
- Struck TH. 2017. Phylogeny of Annelida. In: Gopalakrishnakone P, Malhotra A, editors. Evolution of venomous animals and their toxins. Toxinology. Dordrecht (The Netherlands): Springer.
- Struck TH, Paul C, Hill N, Hartmann S, Hösel C, Kube M, Lieb B, Meyer A, Tiedemann R, Purschke G, et al. 2011. Phylogenomic analyses unravel annelid evolution. Nature. 471(7336):95–98.
- Weigert A, Bleidorn C. 2016. Current status of annelid phylogeny. Org Divers Evol. 16(2):345–362.
- Weigert A, Golombek A, Gerth M, Schwarz F, Struck TH, Bleidorn C. 2016. Evolution of mitochondrial gene order in Annelida. Mol Phylogenet Evol. 94(A):196–206.
- Weigert A, Helm C, Meyer M, Nickel B, Arendt D, Hausdorf B, Santos SR, Halanych KM, Purschke G, Bleidorn C, et al. 2014. Illuminating the base of the Annelid tree using transcriptomics. Mol Biol Evol. 31(6):1391–1401.
- Zhong M, Struck TH, Halanych KM. 2008. Phylogenetic information from three mitochondrial genomes of Terebelliformia (Annelida) worms and duplication of the methionine tRNA. Gene. 416(1–2):11–21.