
Abstract
Cepola schlegelii (Bleeker 1854) belongs to the genus Cepola in the family Cepolidae and order Priacanthiformes. The complete mitochondrial genome of C. schlegelii was sequenced and analyzed by a high-throughput sequencing approach. The full length of the genome is 17,020 bp, including 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNAs), two ribosomal RNA genes (rRNAs), and a non-coding control region (D-loop). Phylogenetic analysis based on complete mitochondrial genomes revealed that C. schlegelii was most closely related to Acanthocepola krusensternii. The complete mitochondrial sequence of C. schlegelii will enrich the mitochondrial genome database and provide useful resources for population genetics and evolution analyses.
Cepola schlegelii is a Eupercaria species found in the South China Sea and the East China Sea (Liu Citation2000; Miyahara et al. Citation2002). Its body is orange-red when it is alive; its back is slightly darker, and its abdomen is slightly lighter. The fins and sides are unmarked, while a black circular spot is visible between the anterior jaw and maxilla. C. schlegelii is a bottom-dwelling fish that inhabits deep sandy or muddy waters. This fish typically digs a cave, hides within it, and stalks its prey by positioning itself near the cave with its heads up and tails down. Molecular genetic studies have not yet been conducted on C. schlegelii. This study provided the first complete mitochondrial genome of this species. Additionally, the evolutionary relationship of Eupercaria fishes was determined through phylogenetic analysis.
The sample was collected from the East China Sea (26°91’N,123°32’E) in October 2021 using the Agassiz trawl method. The voucher specimen (no. FIOSSC01) was stored at the First Institute of Oceanography, Ministry of Natural Resources of China (http://en.fio.org.cn/, Shenghao Liu, [email protected]). Species identification was carried out according to previously described methods (Wu and Zhong Citation2021). Genomic DNA was extracted from the muscle of a dorsal fin using a DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany). Briefly, 25 mg tissue was cut into small pieces and placed in a 1.5 mL microcentrifuge tube. 180 μL ATL buffer and 20 μL Proteinase K were added to the sample and mixed thoroughly by vortexing. The sample was incubated at 56 °C to make the tissue completely lysed. Then, 4 μL RNase A (100 mg/mL) was added to the sample and incubated for 2 min at room temperature (15–25 °C) to remove total RNA. 200 μL Buffer AL was added to the sample, and mix thoroughly by vortexing. Then, 200 μL ethanol (95%) was added and mixed thoroughly by vortexing. The sample mixture was transferred into the DNeasy mini spin column and centrifuge at 10,000 g for 1 min, and the genomic DNA was bound onto the DNeasy membrane. In order to thoroughly wash the genomic DNA, 500 μL Buffer AW1 and Buffer AW2 were added and centrifuged, respectively. The spin column was then air-dried for 3 min at room temperature. Finally, 200 μL Buffer AE was added onto the DNeasy membrane and incubated at room temperature for 2 min, and the genomic DNA was collected by centrifuge for 1 min at 10,000 g.
The mitochondrial genomic DNA was sequenced using the high-throughput sequencing platform MGISEQ-T7 (BGI Genomics, Shenzhen, China) with an insert size of 350 bp. The mitochondrial genome was assembled from clean data with GetOrganelle v1.7.5.3 (Freudenthal et al. Citation2020) according to the reference genome of Acanthocepola krusensternii (GenBank accession number: AP006812). The mitochondrial genome was then annotated using the MitoFish and MITOS databases (Bernt et al. Citation2013; Iwasaki et al. Citation2013). The mitochondrial genome assembly data were deposited in GenBank database from the National Center for Biotechnology Information (NCBI) with accession number ON110125.
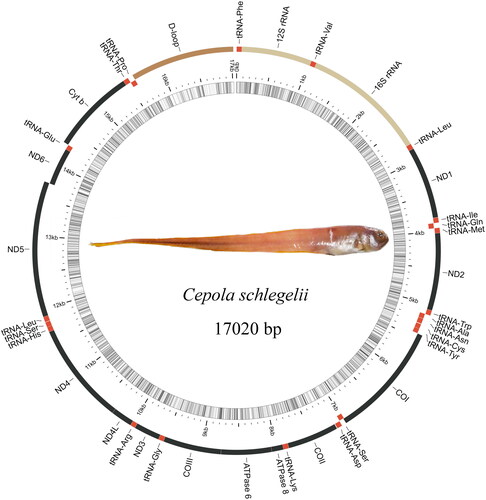
The complete mitochondrial genome is 17,020 bp, containing 13 protein-coding genes (PCGs), 22 transport RNA genes (tRNAs), two ribosomal RNA genes (rRNAs), and one non-coding control region (D-loop) (). The nucleotide composition consists of 26.49% A, 17.27% G, 29.09% C, and 27.15% T. The lengths of the 12S rRNA and 16S rRNA are 949 bp and 1,656 bp, respectively. The sizes of the 22 tRNAs range from 67 bp to 73 bp. PCGs begin with the normal ATG start codon, whereas CO1 starts with GTG. Eight PCGs (ND1, ND2, ND3, ND5, ATP8, ATP6, ND4L, and ND6) terminate with a complete stop codon (ATG/T), whereas three PCGs (CO2, CO3, and ND4) terminate with an incomplete stop codon (T––). The light chain carries eight tRNAs (tRNA-Glu, tRNA-Pro, tRNA-Gln, tRNA-Ala, tRNA-Asn, tRNA-Cys, tRNA-Tyr, and tRNA-Ser) and ND6, while the heavy chain contains the remaining tRNAs.
Figure 1. Mitogenome pattern map of Cepola schlegelii.
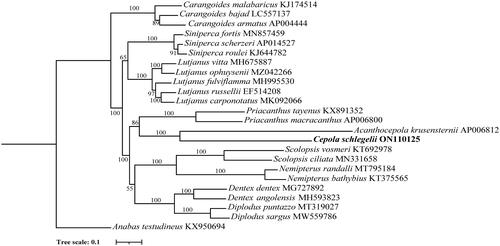
To investigate the evolutionary relationship of C. schlegelii, we constructed a phylogenetic tree using the maximum-likelihood (ML) method based on 13 protein-coding nucleotide sequences from the Eupercaria, Anabantaria and Carangaria mitogenomes. Multiple sequence alignment was conducted using MAFFT (Katoh and Standley Citation2013). Phylogenetic tree was constructed using IQ-TREE software (v1.6.12) from Nucleic acid sequences (with maximum-likelihood method, 1000 replicates, and TVM + F + R5 model; Nguyen et al. Citation2015). The results showed that C. schlegelii and A. krusensternii clustered together, forming the Cepolidae family branch. These two mitochondrial genomes are the only Cepolidae family members in the public database. In the phylogenetic tree, C. schlegelii, A. krusensternii, Priacanthus macracanthus, and Priacanthus tayenus constitute the priacanthiformes order. Diplodus sargus, Dentex dentex, and Scolopsis vosmeri cluster together and form the Spariformes order. Therefore, C. schlegelii is the closest relative to A. krusensternii, while C. schlegelii is far from other species ().
Figure 2. The maximum-likelihood (ML) phylogenetic tree indicated the relationships among Cepola schlegelii and other species in the Eupercaria. Anabas testudineus was used as the outgroup. Numbers on the nodes represent bootstrap values. GenBank accession numbers of each species are listed in the tree.
In conclusion, we presented the complete mitochondrial genome sequence of C. schlegelii and conducted a phylogenetic analysis of Eupercaria. The complete mitochondrial sequence will provide important information for population genetics studies and germplasm conservation.
Ethics statement
The study complied with the ethics of the First Institute of Oceanography, Ministry of Natural Resources.
Author contributions
Panjiao Liang performed the experiments and drafted the manuscript. Shouqiang Wang contributed to genome assembly and annotation. Ye Lin involved in data analysis and interpretation. Li Wang conducted the phylogenetic analysis. Linlin Zhao and Shenghao Liu conceived and supervised the experiments, and revised the manuscript. All authors approved the final version of the manuscript.
Acknowledgments
We would like to thank Mr. Wenge Shi for collecting samples.
Disclosure statement
The authors declare that they have no conflict of interest.
Data availability statement
The complete mitochondrial genome assembly data were available in GenBank database under the accession number ON110125 (https://www.ncbi.nlm.nih.gov/nuccore/ON110125.1/). The high throughput sequencing reads for mitochondrial genome assembly have been deposited in the National Genomics Data Center (NGDC, https://ngdc.cncb.ac.cn) under the BioProject number PRJCA009031, BioSample number SAMC713165, and Genome Sequence Archive (GSA) databse with the accession number CRR458293 (https://ngdc.cncb.ac.cn/gsa/browse/CRA006611/CRR458293).
Additional information
Funding
References
- Bernt M, Donath A, Juhling F, Externbrink F, Florentz C, Fritzsch G, P€utz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319.
- Freudenthal JA, Pfaff S, Terhoeven N, Korte A, Ankenbrand MJ, Förster F. 2020. A systematic comparison of chloroplast genome assembly tools. Genome Biol. 21(1):254.
- Iwasaki W, Fukunaga T, Isagozawa R, Yamada K, Maeda Y, Satoh TP, Sado T, Mabuchi K, Takeshima H, Miya M, et al. 2013. MitoFish and MitoAnnotator: a mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol Biol Evol. 30(11):2531–2540.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Liu M. 2000. Chinese vertebrate encyclopedia. Liaoning: Liaoning University Press.
- Miyahara H, Imamura H, Ishito Y. 2002. Three northern records of the fishes from the pacific coast of aomori prefecture, Japan (Pisces: Teleostei). Bull Fish Sci Hokkaido Univ. 53(1):37–40.
- Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Wu H, Zhong J. 2021. Systematic retrieval of marine and estuarine fish in China. Beijing: China Agriculture Press.