
Abstract
In this study, the complete mitochondrial genome sequence of the New Guinea tiger fish Datnioides campbelli (Whitley 1938) (Lobotiformes: Datnioididae) was sequenced by next-generation sequencing method. The assembled mitochondrial genome consists of 13 protein-coding genes, 22 transfer RNA genes, and two ribosomal RNA genes, with a length of 16,416 bp. The total base composition of the mitogenome of D. campbelli was 29.31% for A, 29.02% for C, 15.14% for G and 26.54% for T. A phylogenetic tree based on 13 protein-coding genes (PCGs) provides important molecular data for further phylogeographic and evolutionary analysis of Lobotiformes.
Introduction
Datnioides campbelli, commonly known as the New Guinea tiger fish, is an ornamental ray-finned fish belonging to the genus Datnioides (Family: Datnioididae, Order: Lobotiformes, Series: Eupercaria). The distribution of the New Guinea tiger fish is limited to the Gulf of Papua drainages and the coastal waters of New Guinea, occurring in brackish river mouths, coastal lagoons, and rivers above tidal influence (Roberts and Kottelat Citation1994). In this study, we focused on the New Guinea tiger fish due to its restricted distribution, high commercial value, and paucity of genetic research. This is the first report about the genomic study on D. campbelli. It provides a novel reference genome and important molecular data for Lobotiformes, which is a fundamental step toward resolving the phylogenetic relationships of the highly diverse ray-finned fish.
Materials
The specimen of D. campbelli () was collected from Guangzhou Lanhai Marine Technology Co., Ltd, Guangzhou city, Guangdong Province, China (latitude: 23°12′51″N, longitude: 113°28′6″E) and identified according to the morphological characters described in Roberts and Kottelat (Citation1994). The muscle was preserved in 95% ethanol and stored at −80 °C. A specimen was deposited at the National Freshwater Genetic Resource Center in Guangzhou city, Guangdong Province in China (https://cafs-germplasm.app.msorg.cn/, Yexin Yang is the contact person [email protected]) under the voucher number DA-camp-1.
Figure 1. The reference picture of Datnioides campbelli in the present study (taken by Yexin Yang).

Methods
Genomic DNA was extracted using a TIANamp Genomic DNA Kit (TIANGEN) and determined by a 0.8% agarose gel and Qubit® 2.0 fluorometer (Life Technologies, USA). The high-quality genomic DNA was used to prepare DNA library with an insert size of 350 bp using NEB Next® Ultra DNA Library Prep Kit for Illumina (NEB, USA) following manufacturer’s recommendations. 2 × 150 bp paired-end reads were generated on an Illumina Novaseq6000 platform using sequencing protocols provided by the manufacturer (Illumina, Inc., San Diego, CA). About 2.07 G raw data was generated. Raw reads from Illumina sequencing were subjected to adaptor trimming and filtering of low-quality reads by fastp v0.20.1(https://github.com/OpenGene/fastp) (Chen et al. Citation2018). The minimum length for reads after trimming was set to 150 nucleotides, and the quality threshold was set to Q20. The whole genome was assembled using SPAdes v.3.15.2 (http://cab.spbu.ru/software/spades/) (Lapidus et al. Citation2014) with ‘–plasmid’ option and kmer sizes 33, 55, 77, 99 and 127. The assembled contigs included a mixture of sequences from organellar and nuclear genomes. We identified mitochondrial contigs using similarity searches by BLASTN 2.13.0+ against NCBI Nucleotide collection (nt) database. MitoFish (http://mitofish.aori.u-tokyo.ac.jp/) (Iwasaki et al. Citation2013) was used to annotate the mitochondrial genome. The circular mitochondrial genome map of D. campbelli was visualized via CGView (Stothard and Wishart Citation2005). The study protocol was approved by the Laboratory Animal Ethics Committee of Pearl River Fisheries Research Institute, CAFS (number: LAEC-PRFRI-20201219).
The complete mitochondrial genome of D. campbelli was blasted against the GenBank database in NCBI, and 17 species highly similar to our D. campbelli, with Max score between 11,924 and 12,381, were selected to perform the phylogenetic analysis, including Banjos banjos (KT345965, Liu et al. Citation2016), Caesio cuning (KP874185, Zhan et al. Citation2017), Datnioides polota (MZ930122, unpublished), Lutjanus argentimaculatus (JN182927, Liao et al. Citation2013), Lutjanus bengalensis (FJ171339, Wang et al. Citation2010), Lutjanus carponotatus (MK092066, Kim et al. Citation2019), Lutjanus fulgens (MN398650, Afriyie et al. Citation2020), Lutjanus kasmira (FJ416614, Wang et al. Citation2010), Lutjanus ophuysenii (MZ042266, Sun et al. Citation2021), Lutjanus peru (KR362299, Bayona-Vásquez et al. Citation2017), Lutjanus rivulatus (AP006000, Yamanoue et al. Citation2007), Lutjanus stellatus (MW485060, unpublished), Monodactylus argenteus (AP009169, Yamanoue et al. Citation2007), Prionurus biafraensis (MN703417, Ludt et al. Citation2020), Prionurus laticlavius (MN703418, Ludt et al. Citation2020), Pterocaesio digramma (LC549803, Song et al. Citation2020), and Stereolepis doederleini (MT083886, Oh et al. Citation2021). Amongst, Monodactylus argenteus was selected as the outgroup. The alignment of concatenated 13 protein-coding genes (PCGs) from our D. campbelli together with the above 17 species were aligned using ClustalW in BioEdit (Hall et al. Citation2011) and then converted to Nexus file by PDGSpider 2.1.1.5 (Lischer abd Excoffier Citation2012), which was used as the import file for later phylogenetic analysis. A Bayesian phylogenetic tree was constructed based on 13 PCGs using MrBayes 3 (Ronquist and Huelsenbeck Citation2003). The specific settings were as follows: Four Metropolis-coupled Markov chain Monte Carlo (MCMC) analyses were run twice for 1,000,000 generations and sampled every 100 generations (mcmc ngen = 1,000,000; nchains = 4; temp = 0.01; samplefreq = 100; burnin = 2500) using the default general-time-reversible + gamma + invariants (GTR + G + I) model of sequence evolution and running until the standard deviation of the split frequencies was below 0.01. Then the first 25% of total trees were removed as burin-in, and the posterior probabilities (PP) was calculated by the remaining trees. The final tree was visualized in FigTree v1.4.4 (http://tree.bio.ed.ac.uk/).
Results
The complete mitochondrial genome of D. campbelli was 16,416 bp in length (GenBank: MZ930121), and included 13 protein-coding genes, 22 transfer RNAs, and two ribosomal RNA genes and a noncoding control region (D-loop) (). The order of the mitogenome of D. campbelli was identical with that of D. polota (MZ930122, unpublished), and the overall sequence identity between D. campbelli and D. polota was up to 84.4%. The overall base composition of the complete mitogenome of D. campbelli was 29.31% for A, 29.02% for C, 15.14% for G, 26.54% for T.
Figure 2. The circular mitochondrial genome map of D. campbelli in this study.
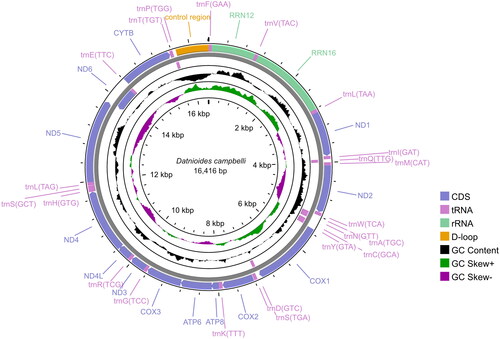
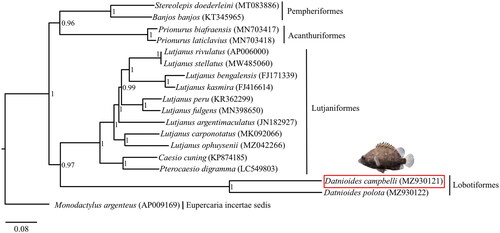
Figure 3. BI phylogenetic tree of 18 species in Eupercaria based on 13 protein-coding genes (PCGs). The numbers beside the nodes represent the Bayesian posterior probability. D. campbelli identified in this study (MZ930121) is labeled with a red box and upside is the picture of this species, taken by Yexin Yang.
The mitogenome of D. campbelli had typical vertebrate organization. All the 13 mitochondrial protein-coding genes shared the same start codon ATG, except for COXI (GTG start codon). The complete stop codon, TAA, was present in ND2, COXI, ATP8, ATP6, COXIII, ND4, ND5 and ND6; TAG was present in ND1, ND3; and the incomplete stop codon ‘T––’ was found in COXII, ND4 and CYTB. Of all protein-coding genes, the longest was the ND5 gene (1,839 bp), and the shortest was the ATP8 gene (168 bp).
Phylogenetic analysis based on 13 PCGs revealed that Lobotiformes was more closely related to Lutjaniformes, compared with Pempheriformes and Acanthuriformes ().
Discussion and conclusion
As the largest and most diverse group of vertebrates, ray-finned fishes account for half of all vertebrate species (Near et al. Citation2012; Hughes et al. Citation2018). In the present study, we first reported a novel mitochondrial genome of D. campbelli and the phylogeny inferred using the Bayesian method based on 13 PCGs showed that Lobotiformes is the sister lineage of Lutjaniformes, which is consistent with the previous study on the phylogeny of ray-finned fishes based on transcriptomic and genomic data (Hughes et al. Citation2018). Our study not only provides critical molecular data for further phylogeographic and evolutionary analysis of Lobotiformes, but also offers a basis for the later deep research on phylogenetic relationships in ray-finned fishes.
Authors’ contributions
Hong Zhou: Performed the experiments, wrote the main manuscript and analyzed the data. Yexin Yang: Collected samples, designed the experiments and analyzed the data. Yi Liu: Performed the experiments and analyzed the data. Hongmei Song: Analyzed the data. Xuejie Wang: Analyzed the data. Sudong Xia: Conceptualization, revised the manuscript. Xidong Mu: Conceptualization, designed the experiments, Funding acquisition. All authors agree to be accountable for all aspects of the work.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for doing the research and writing the paper.
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at (https://www.ncbi.nlm.nih.gov/genbank/) under accession number MZ930121. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA787735, SRR17211917, and SAMN23839792, respectively.
Additional information
Funding
References
- Afriyie G, Wang Z, Dong Z, Ayisi Larbi C, Asiedu B, Guo Y. 2020. Complete mitochondrial genome and assembled DNA barcoding analysis of Lutjanus fulgens (Valenciennes, 1830) and its comparison with other Lutjanus species. Ecol Evol. 10(15):7971–7980.
- Bayona-Vásquez NJ, Hernández-Álvarez CA, Glenn T, Domínguez-Domínguez O, Uribe-Alcocer M, Díaz-Jaimes P. 2017. Complete mitogenome sequences of the pacific red snapper (Lutjanus peru) and the spotted rose snapper (Lutjanus gutattus). Mitochondrial DNA Part A. 28(2):223–224.
- Chen S, Zhou Y, Chen Y, Gu J. 2018. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 34(17):i884–i890.
- Hall T, Biosciences I, Carlsbad CJGBB. 2011. BioEdit: an important software for molecular biology. GERF Bulletin of Biosciences. 2(1):60–61.
- Hughes LC, Ortí G, Huang Y, Sun Y, Baldwin CC, Thompson AW, Arcila D, Betancur-R R, Li C, Becker L, et al. 2018. Comprehensive phylogeny of ray-finned fishes (Actinopterygii) based on transcriptomic and genomic data. Proc Natl Acad Sci USA. 115(24):6249–6254.
- Iwasaki W, Fukunaga T, Isagozawa R, Yamada K, Maeda Y, Satoh TP, Sado T, Mabuchi K, Takeshima H, Miya M, et al. 2013. MitoFish and MitoAnnotator: a mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol Biol Evol. 30(11):2531–2540.
- Kim G, Lee JH, Alam MJ, Lee SR, Andriyono S. 2019. Complete mitochondrial genome of Spanish flag snapper, Lutjanus carponotatus (Perciformes: lutjanidae). Mitochondrial DNA B. 4(1):568–569.
- Lapidus A, Antipov D, Bankevich A, Gurevich A, Korobeynikov A, Nurk S, Prjibelski A, Safonova Y, Vasilinetc I, Pevzner PA. 2014. New frontiers of genome assembly with SPAdes 3.0 [poster]. http://cab.spbu.ru/software/spades/.
- Liao J, Wang ZD, Guo YS, Liu CW. 2013. Complete mitochondrial sequence of Lutjanus argentimaculatus and Bayesian analysis. Marine Sciences. 37(1):62–69.
- Lischer HE, Excoffier L. 2012. PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics. 28(2):298–299.
- Liu J, Jin W, Wu C. 2016. Complete mitochondrial genome of banjofish (Banjos banjos): genome characterization and phylogenetic analysis. Mitochondrial DNA A. 27(6):4433–4435.
- Ludt WB, Rocha LA, Chakrabarty P. 2020. The first complete mitochondrial genomes of sawtail surgeonfishes (Acanthuridae: prionurus). Mitochondrial Part B. 5(1):212–213.
- Near TJ, Eytan RI, Dornburg A, Kuhn KL, Moore JA, Davis MP, Wainwright PC, Friedman M, Smith WL. 2012. Resolution of ray-finned fish phylogeny and timing of diversification. Proc Natl Acad Sci USA. 109(34):13698–13703.
- Oh DJ, Lee JC, Ham YM, Jung YH. 2021. The mitochondrial genome of Stereolepis doederleini (Pempheriformes: polyprionidae) and mitogenomic phylogeny of Pempheriformes. Genetics and Molecular Biology. Genet Mol Biol. 44(1):e20200166.
- Roberts TR, Kottelat M. 1994. The Indo-Pacific tigerperches, with a new species from the Mekong basin (Pisces: coiidae). Ichthyol Explor Freshwaters. 5: 257–266.
- Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 19(12):1572–1574.
- Song HY, Jung YH, Kim B, Choi YJ, Nguyen TV, Lee DS. 2020. Complete mitochondrial genome of the double-lined fusileer, Pterocaesio digramma (Perciformes, Caesionidae): mitogenome characterization and phylogenetic analysis. Mitochondrial DNA B Resour. 5(3):2617–2618.
- Stothard P, Wishart DS. 2005. Circular genome visualization and exploration using CGView. Bioinformatics. 21(4):537–539.
- Sun P, Jiang Y, Yuan X, Zhang H. 2021. The complete mitochondrial genome of Lutjanus ophuysenii and phylogenetic analysis. Mitochondrial DNA B Resour. 6(8):2396–2397.
- Wang ZD, Tan W, Guo YS, Liu L, Liu CW, Liu Y. 2010. Performance of mitogenomic coding regions within genus Lutjanus molecular phylogenetics. J Fish China. 34(6):656–664.
- Yamanoue Y, Miya M, Matsuura K, Yagishita N, Mabuchi K, Sakai H, Katoh M, Nishida M. 2007. Phylogenetic position of tetraodontiform fishes within the higher teleosts: Bayesian inferences based on 44 whole mitochondrial genome sequences. Mol Phylogenet Evol. 45(1):89–101.
- Zhan W, Chen RY, Shen KN, Xu DD, Hsiao CD, Lou B. 2017. Next-generation sequencing yields the complete mitochondrial genome of the Redbelly yellowtail fusilier, Caesio cuning (Teleostei: caesionidae). Mitochondrial DNA A. 28(1):125–126.