
Abstract
Epiverta chelonia (Mader 1933; Coleoptera: Coccinellidae) is an important economically and scientifically valuable insect. In this study, the first complete mitochondrial genome of E. chelonia was sequenced and characterized using next-generation sequencing techniques. The circular mitogenome of E. chelonia consists of 17,347 bp including 13 protein-coding genes (PCGs), 22 transfer RNA (tRNA) genes, two ribosomal RNA (rRNA) genes, and a control region (D-loop). The base composition was AT-biased (75.77%). Bayesian Inference and Maximum likelihood phylogenetic trees strongly supported the monophyly of Coccinellinae. Also, E. chelonia was supported as the sister group of Subcoccinella vigintiquatuorpunctata, within Epilachninae. Thus, the E. chelonia mitochondrial genome will be a fundamental resource for understanding the molecular phylogenetic relationships of the species-rich family Coccinellidae of Coleoptera.
Epiverta chelonia (Mader Citation1933) belongs to the Coccinellid tribe Epivertini (Mader Citation1933; Dieke Citation1947), and is a medium to large-sized lady beetle. Its body is regularly oval, length is 4.6–8.1 mm, and width is 4.1–6.4 mm. The surface of the elytra is distinctly brownish-black with regular yellow maculae. Also, the pubescence is moderately dense, long, and yellowish. E. chelonia is distributed in southwestern China including the southwestern Sichuan Province, and northwestern Yunnan Province (Pang and Mao 1979; Katoh et al. Citation2014; Szawaryn et al. Citation2015; Tomaszewska et al. Citation2017). It mainly feeds on the flowers, leaves, and stems of the Anemone spp. (Ranunculaceae) and Artemisia sp. (Asteraceae) (Pang and Mao 1979). It was listed in the “Lists of terrestrial wildlife under state protection, which are beneficial or of important economic or scientific value” by the National Forestry Administration on 1 August 2000 (https://www.forestry.gov.cn/main/3954/content-959027.html). Therefore, the complete mitochondrial genome of E. chelonia was analyzed to provide new insights into the phylogeny of Coccinellidae.
In this study, all the specimens were collected from the leaves of Aconitum carmichaelii Debeaux in Heqing County, Dali Bai Autonomous Prefecture, Yunnan Province, China (26° 28′32.43″N, 100° 4′3.24″E) in 2021 and were subsequently identified to species using morphology. Ethics approval was not applicable for this study because it did not contain any human participants or vertebrates. The voucher specimens were deposited in the Plant Protection College, Yunnan Agricultural University (https://www.ynau.edu.cn/, Yongke Zhang, [email protected]) under the voucher number GPC-HQ-1. The total DNA was extracted from six E. chelonia individuals, which were collected from the same site and host plant, using the CTAB method. Mitogenome sequencing was performed using the Illumina NovaSeq 6000 (Biomarker Biotechnology Co., Ltd, Beijing, China). Raw and clean reads were assembled using the program Getorganelle v. 1.7.1a (Jin et al. Citation2020). Next, the assembled contigs were aligned with the mitochondrial genome of related species using blastn (BLAST 2.2.30+). Then, the MITOS web and CGView servers were used for gene annotation and mapping of the graphic view of the mitogenomes, respectively (Grant and Stothard 2008; Bernt et al. Citation2013).
The complete mitochondrial genome of E. chelonia was submitted to Genbank (accession number ON209194). It was a closed circular molecule of 17,347 bp in length containing 13 protein-coding genes (PCGs), 22 transfer RNA (tRNA) genes, two ribosomal RNA (rRNA) genes, and a control region (D-loop). The nucleotide composition was AT-biased (A: 39.25%, C: 15.03%, G: 9.19%, and T: 36.52%). The total length of the 22 tRNAs was 1415 bp, with sizes ranging from 55 bp (trnS1) to 70 bp (trnK); the A + T content ranged from 56.92% (trnI) to 90.32% (trnE). Also, the two rRNA genes were 1270 bp (rrnL) and 813 bp (rrnS) in length, and the 13 PCGs had an overall length of 11,052 bp. Additionally, four PCGs (nad1, nad4, nad4l, and nad5) were encoded by the minority strand (N-strand) while the other nine were located on the majority strand (J-strand). All the tRNAs in E. chelonia were predicted to fold into a typical cloverleaf secondary structure, except for three genes (trnH, trnL1, and trnY) and one gene (trnS1), which lacked a TψC loop and dihydrouracil arm (DHU arm) and loop, respectively, and therefore, could not fold into a typical cloverleaf structure. The 16 s rRNA (1270 bp) was located between trnL1 and trnV, and the 12S rRNA (813 bp) resided between trnV and D-loop. Their A + T contents were 81.10% and 77.61%, respectively. All the PCGs of E. chelonia had the conventional initiation codon for invertebrate mitochondrial PCGs (ATN), except for cox1 (TTG). Eleven and two of the PCGs stopped with the termination codons TAA (atp6, atp8, cob, cox1, cox2, cox3, nad2, nad4, nad4l, nad5, nad6) and TAG (nad1, nad3), respectively.
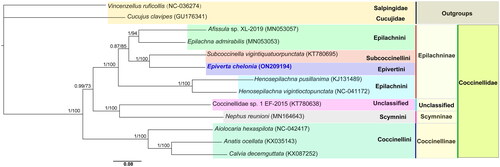
The phylogenetic analysis was carried out using 13 PCGs of E. chelonia and 12 other species (10 Coccinellidae, 1 Salpingidae and 1 Cucujidae). Additionally, two species from Salpingidae and Cucujidae were chosen as outgroups. The 13 PCG sequences were aligned using the MAFFT algorithm in TranslatorX. The partitions and models were estimated using PartitionFinder 2 (Lanfear et al. Citation2017). Then the maximum likelihood analyses and Bayesian inference analyses were conducted with IQ-TREE (Nguyen et al. Citation2015) and MrBayes v. 3.2.6 (Ronquist et al. Citation2012) in PhyloSuite (Zhang et al. Citation2020), respectively. The two phylogenetic analyses using different methods yielded the same topology, with only some nodal supporting values which were different. The monophyly at the tribe level within the subfamily was strongly supported in the phylogenetic trees. Also, E. chelonia as a sister group to Subcoccinella vigintiquatuorpunctata was well supported (). In terms of the topology, the species could be divided into three clades from the consensus trees: Epilachninae coccinellids clustered together to form the first clade branching from the base of the tree; Scymnini coccinellids and Coccinellidae sp. 1 EF-2015 (unclassified) clustered together to form the second clade; and Coccinellini coccinellids clustered together to form the third clade. These results provide an important basis for further studies on the mitochondrial genome and phylogenetics of Coccinellidae.
Figure 1. The phylogram was constructed using Bayesian inference (BI) analyses by 2 million generations were run, with 25% of the generations as burn-in. PSRF close to 1.0 and standard deviation of split frequencies below 0.01 were accepted. BI (posterior probabilities) and ML (bootstrap values) support values are reported above nodes, respectively.
Ethical approval
The study does not involve the study of vertebrates or regulated invertebrates, and samples are collected in areas available for collection in public areas, in accordance with guidelines provided by the authors’ institutions and national or international regulations. Therefore, ethical approval is not required for this study.
Authors’ contributions
Yongke Zhang: Conceptualization, Investigation, Data Curation, Formal analysis, Writing – Original Draft; Lijuan Zhang: Data Curation, Formal analysis, Writing – Review and Editing; Yingchun Lu: Investigation, Resources, Data Curation; Xiahong He: Funding Acquisition, Supervision; Hongrui Zhang (Corresponding Author): Samples collection, Funding Acquisition, Supervision, Visualization, Writing – Review and Editing, Project administration. All authors agree to be accountable for all aspects of the work.
Acknowledgments
We would like to thank Researcher Guoyue Yu for identifying the species and Dr. Shengchang Lai for the data analysis.
Disclosure statement
The authors report no conflict of interest. The authors are responsible for the sample collection, experiment designing, writing, and revising of the paper.
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov/ under the accession no. ON209194. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA847008, SRR19612121, and SAMN28928437, respectively.
Additional information
Funding
References
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319.
- Dieke GH. 1947. Ladybeetles of the genus Epilachna (Sens. Lat) in Asia, Europe, and Australia. Smithsonian Miscellaneous Collect. 106(15):1–183.
- Grant JR, Stothard P. 2008. The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 36(Web Server issue):W181–W184.
- Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. 2017. Partitionfinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol. 34(3):772–773.
- Jin JJ, Yu WB, Yang JB, Song Y, dePamphilis CW, Yi TS, Li DZ. 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):241.
- Katoh T, Koji S, Ishida TA, Matsubayashi KW, Kahono S, Kobayashi N, Furukawa K, Viet BT, Vasconcellos-Neto J, Lange CN, et al. 2014. Phylogeny of Epilachna, Henosepilachna, and some minor genera of phytophagous ladybird beetles (Coleoptera: Coccinellidae: Coccinellinae: Epilachnini), with an analysis of ancestral biogeography and host-plant utilization. Zoolog Sci. 31(12):820–830.
- Mader L. 1933. Über bekannte und neue coccinelliden. Entomologischer Anzeiger. 13:79–84.
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Pang XF, Mao JL. 1979. Economic insect fauna of China (XIV). In: Coleoptera: Coccinellidae II. Beijing: Science Press; p. 158–160. (In Chinese).
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61(3):539–542.
- Szawaryn K, Bocak L, ŚlipiŃski A, Escalona HE, Tomaszewska W. 2015. Phylogeny and evolution of phytophagous ladybird beetles (Coleoptera: Coccinellidae: Epilachnini), with recognition of new genera. Syst Entomol. 40(3):547–569.
- Tomaszewska W, Huo LZ, Szawaryn K, Wang XM. 2017. Epiverta Dieke (Coleoptera: Coccinellidae: Epilachnini): a complex of species, not a monotypic genus. J Insect Sci. 17(2):1–12.
- Zhang D, Gao FL, Jakovlic I, Zou H, Zhang J, Li WX, Wang GT. 2020. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour. 20(1):348–355.