
Abstract
Lophostemon confertus (Myrtaceae), a fast-growing ornamental tree, is widely cultivated in tropical and subtropical regions. To determine its phylogenetic position within Myrtaceae, here we report its complete chloroplast (cp) genome, which is 160,297 bp long and contains two inverted repeats (IRs) of 26,490 bp each, separated by a small single-copy region of 18,826 bp and a large single-copy region of 88,491 bp. The cp genome contains 123 genes, including 73 unique protein-coding genes (six duplicated in the IR regions), 29 unique tRNA genes (seven duplicated in the IR regions), and four unique rRNA genes (all located in the IR regions). Phylogenetic analysis of 18 species of Myrtaceae showed that L. confertus is sister to Xanthostemon chrysanthus. The complete cp genome of L. confertus provides a valuable genetic resource for further phylogenetic studies.
1. Introduction
Lophostemon confertus (Br.) Peter G. Wilson & J. T. Waterhouse (Citation1982), a fast-growing and popular ornamental tree, is native to Australia, and is widely cultivated in tropical and subtropical regions. Due to its rapid growth, evergreen leaves, and air pollution resistance, it is used as a multipurpose tree, from an ideal landscape resource in scenic zones and city roads to excellent pioneer species for wasteland afforestation. Moreover, the superior quality of its timber, like hard, moisture-proof and termite-resistant, endows it with important economic value, and becomes an important economic tree in China. Sufficient genome resources can not only do great help to species identification and phylogenetic analysis, but also provide a molecular basis for marker-assisted breeding and horticultural variety improvement. However, little genetic and genomic information is available for this species, and more generally, even neither for the tribe Leptospermoneae to which it belongs. In this study, the complete chloroplast of L. confertus was first assembled through genome skimming sequencing data and its phylogenetic position within Myrtaceae was resolved.
2. Materials and methods
The leaves and fruits of Lophostemon confertus were photographed by ourselves and shown in . This species is not an endangered or protected species, so no permissions are required to obtain the sample. Fresh young leaves of an individual L. confertus were collected from the campus of Sun Yat-sen University (N113°18'8″, E23°5'24″), Guangzhou, China. A voucher specimen was identified by Rifan Liang and deposited at Sun Yat-sen University Herbarium (SYS) (http://cfh.ac.cn/subsite/default.aspx?siteid=SYS) with the specimen voucher code SYS-Bore-2021-10–13 under the charge of Wenbo Liao ([email protected]). Total DNA was extracted from fresh leaves using the CTAB method (Doyle and Doyle Citation1987). Genomic DNA was fragmented into ∼350 bp fragments by an ultrasonic processor to construct a DNA library, which was then sequenced on an Illumina NovaSeq 6000 platform. Approximately 5.0 Gbp of paired-end sequences (2 × 150 bp) were generated and filtered using Trimmomatic (Bolger et al. Citation2014). Clean reads were used to assemble the chloroplast genome using GetOrganelle v1.6.4 with default parameters (Jin et al. Citation2020) by de novo algorithm without a reference genome. To clarify the accuracy of the assembly, we further mapped our clean reads back to the assembled chloroplast (cp hereafter) genome to assess the depth of coverage (Figure S1). Culminated in stitching into a synthetic loop, the chloroplast genome was automatically annotated by a web server CPGAVAS2 (Shi et al. Citation2019) and double-checked by Geneious version 11.1.5 (Kearse et al. Citation2012) followed by manual adjustment and confirmation. The structures of intron-containing genes were visualized in CPGVIEW (http://www.1kmpg.cn/cpgview, unpublished). OGDRAW (Lohse et al. Citation2013) was applied to visualize the gene map of L. confertus (). To infer the phylogenetic position of L. confertus in Myrtaceae, the complete cp genome sequences of 18 species in this family (including L. confertus) were used for phylogenetic reconstruction. Two species each from Melastomataceae and Lythraceae were used as the outgroups. All the complete cp genome sequences of these species except L. confertus were downloaded from GenBank with their accession numbers shown in . After sequence alignment with MAFFT v7.307 (Katoh and Standley Citation2013), a maximum likelihood (ML) tree was constructed based on the GTRGAMMA substitution model with 1000 bootstrap replicates using RAxML v8.2.12 (Stamatakis Citation2014).
Figure 1. A branch of Lophostemon confertus showing the morphology of leaves and fruits. Photographed by Wenhang Su.

Figure 2. The chloroplast genome map of Lophostemon confertus. Genes are shown outside and inside the outer circle are transcribed counterclockwise and clockwise, respectively. GC and AT contents across the chloroplast genome are shown with dark and light shading, respectively, inside the inner circle.
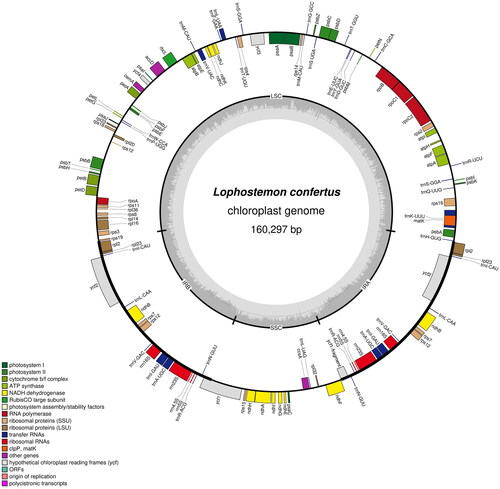
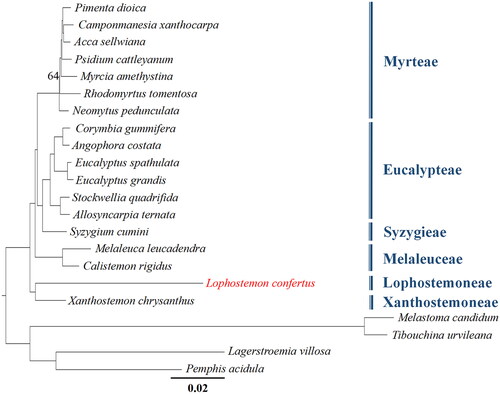
Figure 3. Phylogenetic tree based on the complete chloroplast genome sequences of 18 species from Myrtaceae, with four species from Melastomateae and Lythraceae, used as outgroups. Bootstrap support values of all but one node are 100 (omitted here), and only one node with a bootstrap support value of 64 is shown. The following sequences were used: Pimenta dioica KY085891.1 (unpublished), Camponmanesia xanthocarpa KY392760.1 (unpublished), Acca sellwiana KX289887.1 (Machado et al. Citation2017), Psidium cattleyanum MN095413.1 (Rodrigues et al. Citation2020), Myrcia amethystina MW353255.1 (Lima et al. Citation2021), Rhodomyrtus tomentosa MK044696.1 (Huang et al. Citation2019), Neomytus pedunculata MW214671.1 (Maurin et al. Citation2022), Corymbia gummifera KC180800.1 (Bayly et al. Citation2013), Angophora costata KC180805.1 (Bayly et al. Citation2013), Eucalyptus spathulata KC180793.1 (Bayly et al. Citation2013), Eucalyptus grandis MT700491.1 (unpublished), Stockwellia quadrifida KC180807.1 (Bayly et al. Citation2013), Allosyncarpia ternata KC180806.1 (Bayly et al. Citation2013), Syzygium cumini GQ870669.3 (unpublished), Melaleuca leucadendra MT700493.1 (unpublished), Calistemon rigidus MN794317.1 (Liu et al. Citation2020), Xanthostemon chrysanthus MW837774.1 (unpublished), Melastoma candidum KY745894.1 (Ng et al. Citation2017), Tibouchina urvileana NC043810.1 (Gonçalves et al. Citation2019), Lagerstroemia villosa MK881633.1 (Gu et al. Citation2019), Pemphis acidula MH727532.1 (Jian and Ren Citation2019).
3. Results
The complete cp genome of L. confertus (GenBank accession No. OM640421.1) was 160,297 bp long, with an overall GC content of 36.81%. It contained two inverted repeats (IRs) of 26,490 bp each, separated by an 18,826 bp small single-copy (SSC) region and an 88,491 bp large single-copy (LSC) region. It has 123 genes, including 73 unique protein-coding genes (six duplicated in the IR regions), 29 unique tRNA genes (seven duplicated in the IR regions) and four unique rRNA genes (all located in the IR regions), in which rps12 and ycf3 had two introns (Figure S2). Like other angiosperms, L. confertus exhibits trans-splicing in rps12.
As shown in the phylogenetic tree (), the relationships of the six tribes of Myrtaceae with available cp genome data are well resolved, and L. confertus aligns as a sister group to Xanthostemon chrysanthus, both with high bootstrap support.
4. Discussion and conclusion
The cp genome of Lophostemon confertus (Myrtaceae), a fast-growing roadside tree, was first sequenced and assembled in this study. Phylogenetic analysis of 18 species of Myrtaceae showed that L. confertus, which belongs to the tribe Leptospermoneae, is sister to Xanthostemon chrysanthus from another tribe Xanthostemoneae.
Myrtaceae, consisting of 17 tribes and about 5650 species, is one of the largest families of flowering plants (Wilson et al. Citation2005; Govaerts et al. Citation2015). The phylogeny of this family has attracted much attention, but most of the studies were conducted based on the use of very few molecular markers, such as nuclear ribosomal internal transcribed spacers (nrITS) (Lucas et al. Citation2007; Biffin et al. Citation2010), matK (Wilson et al. Citation2005) and ycf2 (Machado et al. Citation2020). Recent phylogenetic analyses based on complete cp genome sequences have been performed mainly from the species-rich tribes Myrteae, Eucalypteae and Syzygieae (Bayly et al. Citation2013; Machado et al. Citation2017; Machado et al. Citation2020; Balbinott et al. Citation2022). To date, no species from the tribe Lophostemoneae has available cp genome sequences. The complete cp genome of L. confertus provides a valuable genetic resource for comprehensively resolving the phylogenetic relationships among 17 tribes of Myrtaceae in near future.
Ethical approval
No ethical issues were involved in this study. The collection of plant sample was legal and reasonable. A voucher specimen has been identified by Rifan Liang and deposited at Sun Yat-sen University Herbarium (SYS) with the specimen voucher code SYS-Bore-2021-10-13 under the charge of Wenbo Liao ([email protected]). Information on the voucher specimen and who identified it were included in the manuscript.
Author contributions
Wenhang Su and Rifan Liang designed the study. Wenhang Su was in charge of material collection, data analysis and manuscript drafting. Rifan Liang revised the manuscript. These two authors agree to be accountable for all aspects of the work.
Supplemental Material
Download PNG Image (251.4 KB){kind=link}
Supplemental Material
Download PNG Image (392.3 KB){kind=link}
Disclosure statement
No potential competing interest was reported by the authors.
Data availability statement
The data that support the findings of this study are openly available in NCBI at https://www.ncbi.nlm.nih.gov/OM640421 under the accession no. OM640421. And the associated Bioproject, SRA, Bio-sample numbers are PRJNA804362, SRR17931549 and SAMN25731117, respectively
Additional information
Funding
References
- Balbinott N, Rodrigues NF, Guzman FL, Turchetto-Zolet AC, Margis R. 2022. Perspectives in Myrtaceae evolution from plastomes and nuclear phylogenies. Genet Mol Biol. 45(1):e20210191.
- Bayly MJ, Rigault P, Spokevicius A, Ladiges PY, Ades PK, Anderson C, Bossinger G, Merchant A, Udovicic F, Woodrow IE, et al. 2013. Chloroplast genome analysis of Australian eucalypts – Eucalyptus, Corymbia, Angophora, Allosyncarpia and Stockwellia (Myrtaceae). Mol Phylogenet Evol. 69(3):704–716.
- Biffin E, Lucas EJ, Craven LA, Ribeiro da Costa I, Harrington MG, Crisp MD. 2010. Evolution of exceptional species richness among lineages of fleshy-fruited Myrtaceae. Ann Bot. 106(1):79–93.
- Bolger AM, Marc L, Bjoern U. 2014. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 30(15):2114–2120.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Gonçalves DJP, Simpson BB, Ortiz EM, Shimizu GH, Jansen RK. 2019. Incongruence between gene trees and species trees and phylogenetic signal variation in plastid genes. Mol Phylogenet Evol. 138(29):219–232.
- Govaerts R, Sobral M, Ashton P, Barrie F, Holst BK, Landrum LL, Matsumoto K, Mazine FF, Lughadha EN, Proenca C, et al. 2015. World checklist of Myrtaceae. Kew: Royal Botanic Gardens.
- Gu C, Ma L, Wu Z, Chen K, Wang Y. 2019. Comparative analyses of chloroplast genomes from 22 Lythraceae species: inferences for phylogenetic relationships and genome evolution within Myrtales. BMC Plant Biol. 19(1):1–19.
- Huang Y, Yang Z, Huang S, An W, Li J, Zheng X. 2019. Comprehensive analysis of Rhodomyrtus tomentosa chloroplast genome. Plants. 8(4):89.
- Jian S, Ren H. 2019. The complete chloroplast genome sequence of Pemphis acidula (Lythraceae). Mitochondrial DNA Part B. 4(1):912–913.
- Jin J-J, Yu W-B, Yang J-B, Song Y, dePamphilis CW, Yi T-S, Li D-Z. 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):241.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Lima DF, Goldenberg R, Forest F, Cowan RS, Lucas EJ. 2021. Phylogeny and biogeography of Myrcia sect. Aguava (Myrtaceae, Myrteae) based on phylogenomic and sanger data provide evidence for a cerrado origin and geographically structured clades. Mol Phylogenet Evol. 157(3):107043.
- Liu F, Movahedi A, Yang W, Xu L, Xie J, Zhang Y. 2020. The complete chloroplast genome and characteristics analysis of Callistemon rigidus R.Br. Mol Biol Rep. 47(7):5013–5024.
- Lohse M, Drechsel O, Kahlau S, Bock R. 2013. OrganellarGenomeDRAW – a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41:575–581.
- Lucas EJ, Harris SA, Mazine FF, Belsham SR, Nic Lughadha EM, Telford A, Gasson PE, Chase MW. 2007. Suprageneric phylogenetics of Myrteae, the generically richest tribe in Myrtaceae (Myrtales). Taxon. 56(4):1105–1128.
- Machado LdO, Vieira LdN, Stefenon VM, Faoro H, Pedrosa FdO, Guerra MP, Nodari RO. 2020. Molecular relationships of Campomanesia xanthocarpa within Myrtaceae based on the complete plastome sequence and on the plastid ycf2 gene. Genet Mol Biol. 43(2):e20180377.
- Machado LDO, Vieira LDN, Stefenon VM, Pedrosa FDO, Souza EMD, Guerra MP, Nodari RO. 2017. Phylogenomic relationship of feijoa (Acca sellowiana (O.Berg) Burret) with other Myrtaceae based on complete chloroplast genome sequences. Genetica. 145(2):163–174.
- Maurin K, Smissen RD, Lusk CH. 2022. A dated phylogeny shows Plio-Pleistocene climates spurred evolution of anti-browsing defences in the New Zealand flora. New Phytol. 233(1):546–554.
- Ng WL, Cai Y, Wu W, Zhou R. 2017. The complete chloroplast genome sequence of Melastoma candidum (Melastomataceae). Mitochondrial DNA Part B. 2(1):242–243.
- Rodrigues NF, Balbinott N, Paim I, Guzman F, Margis R. 2020. Comparative analysis of the complete chloroplast genomes from six neotropical species of Myrteae (Myrtaceae). Genet Mol Biol. 43(2):e20190302.
- Shi L, Chen H, Jiang M, Wang L, Wu X, Huang L, Liu C. 2019. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47(W1):65–73.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Wilson PG, O’Brien MM, Heslewood MM, Quinn CJ. 2005. Relationships within Myrtaceae sensu lato based on a matK phylogeny. Plant Syst Evol. 251(1):3–19.
- Wilson PG, Waterhouse JT. 1982. A review of the genus Tristania R. Br. (Myrtaceae): a heterogeneous assemblage of five genera. Aust J Bot. 30(4):413–446.