
Abstract
The mitochondrial genome of Camellia nitidissima was sequenced by Illumina and Pacbio sequencing. The results of sequences showed that a total length was 949,915 bp, and the GC content was 45.7% in assembled mitochondrial genome of C. nitidissima. 71 unigenes had been found, including 36 coding proteins and 35 non-coding proteins. Subsequently, the phylogenetic tree was built on 24 plants with the maximum-likelihood method, which had high bootstrap value and fited to the angiosperm phylogeny group classification (APG IV). The study’s findings unravel the taxonomic status of C. nitidissima and benefit the evolution study.
Introduction
Camellia nitidissima C.W.Chi (1948), with golden flowers, belongs to Camellia genus of the family Theaceae, and it is mainly distributed in Guangxi, China. Camellia nitidissima is a kind of Chinese herbal medicine, whose flowers and leaves are particularly rich in saponins, flavonoids and polysaccharides (Hou et al. Citation2018), and C. nitidissima can be used in treatment of dysentery, hypertension, pharyngitis and hematochezia (Wang et al. Citation2016; He et al. Citation2018). Recently, the studies of C. nitidissima mainly focus on the formation of flower color (Zhou et al. Citation2017; Li et al. Citation2019), the types of secondary compounds (Jiang et al. Citation2020), and the extraction of chemical substances (Lin et al. Citation2013), but the mitochondrial genome of C. nitidissima has not been reported. However, the mitochondrial genome of C. nitidissima benefits to clarify its evolutionary position, so we propose to sequence and assemble the complete mitochondrial genome of C. nitidissima, and provide valuable genomic information for phylogeny.
Materials
Camellia nitidissima is a shade-tolerant species and prefers warm, humid climate and acid soil with good drainage, and it is mainly propagated by seeds and cuttings. Camellia nitidissima were cultivated in the nursery of Yulin Normal University (N 22°40′58″, E 110°11′27″), Guangxi, China (). The purple leaves of 7-year-old C. nitidissima plants were collected, cleaned, immediately frozen in liquid nitrogen, and stored at −80 °C. The studied specimen and genomic DNA of C. nitidissima were stored in the Herbarium of Yulin Normal University (https://syy.ylu.cn/index.html, Yulin Zhu, [email protected]) under the voucher number YLU20210011.
Figure 1. Morphology features of C. nitidissima. (a) Individual of C. nitidissima; (b) Mature leaves; (c) Buds and flowers. The photos of C. nitidissima were taken at the nursery of Yulin normal university, Yulin, Guangxi, China.

Methods
Mitochondria of C. nitidissima was isolated from purple and young leaves with the density gradient centrifugation, and the contamination of genomic DNA was eliminated by DNase I (Promega, Madison, USA). DNA quality of mitochondria was checked by Qubit fluorometer (Thermo, Massachusetts, USA) and agarose gel electrophoresis. The sequencing library for Illumina was constructed using the NEBNext® Ultra™ DNA Library Prep Kit (New England Biolabs, Suffolk, England) and sequenced by Illumina NovaSeq 6000 (Illumina, San Diego, USA). The SMRTbell libraries for Pacbio was constructed using the Express Template Prep Kit 2.0 (Pacific Biosciences, California, USA) according to the manufacturer’s protocol and sequenced by Pacbio Sequel II (Pacific Biosciences, California, USA). All of the above were entrusted to Biozeron company (Biozeron, Shanghai, China).
We used two strategies to assemble the mitogenome of C. nitidissima. In the first strategy, the short clean reads were de novo assembled with GetOrganelle v1.6.4 (parameters: -k 21, 65, 105) (Jin et al. Citation2020). Then, in order to extract the potential mitochondrial contigs, the assembled mitochondrial protein-coding genes were alignment with the plant mitogenome database by BLAST. Subsequently, the Pacbio long mitochondrial reads, which were used as bait, were mapped to the potential mitochondrial contigs by BLASR v5.1 with default parametersand assembled by Canu v2.1.1 (parameters: genomeSize = 140m, rawErrorRate = 0.3, correctedErrorRate = 0.045, corOutCoverage = 30) (Koren et al. Citation2017). In the second strategy, firstly, all Pacbio long reads were de novo assembled to get the draft contigs with Canu v2.1.1. Secondly, the short clean reads were mapped to the draft contigs by using BWA (parameters: bwa mem -t 4), and the draft contigs were improved by Pilon v1.22 (parameters: –fix all). Then, MUMmer 3.23 was used to check whether these contigs were circular. And finally, two corrected contigs, which were obtained from two assembly strategies, were align to each other by using MUMmer (parameters: nucmer –prefix). If these two contigs were identical, a master circle of the C. nitidissima mitogenome was correctly assembled. Mitochondrial genes of C. nitidissima were annotated by the GeSeq tool (Tillich et al. Citation2017), with the default parameters, to predict coding proteins, tRNA, and rRNA. And then those genes of functional annotations were performed to blast against (evalue < 1e-10) the non-redundant protein database (Nr), Swiss-Prot, Clusters of Orthologous Groups (COGs), and Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology terms (GO). Subsequently the position of each coding gene was determined using BLAST searches against ref mitochondrion genes in NCBI. The mitochondrial genome map was drawn by the OGDRAW tool. Based on 23 conserved protein-coding genes in the mitochondrial genomes, phylogenetic tree of C. nitidissima and 23 plants was constructed with maximum-likelihood estimation by PhyloSuite software with the best-fit model (GTR + F+R2) and 1000 bootstrap replicates (Zhang et al. Citation2020).
Results
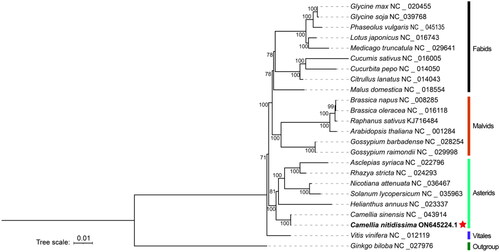
The sequenced results showed the 94.1 million reads and 14.1 Gb raw data were obtained by two sequencing technologies. The assembled mitochondrial genome of C. nitidissima was a single circle with a total length of 949,915 bp, and the GC content was 45.7%. In total, mean sequencing depth was 1044.91 ×(Figure S1, Table S1). There has been found 36 coding proteins and 35 non-coding proteins, including 30 transfer RNA (tRNA), 5 ribosomal RNA (rRNA), in the mitochondrial genome of C. nitidissima. The total length of coding proteins and non-coding proteins were 30,051 and 5,687 bp, accounting for 3.16 and 0.599% of the total genome length, respectively. 36 protein-coding proteins contained electron chain complex of oxidative phosphorylation (16), cytochrome c biosynthesis (6), ribosomal proteins (12), maturases (1) and transport membrane protein (1). In addition, 9 genes, such as ccmFc, ccmFn, cob, nad3, rpl5, rps12, rps14, rrnS and rrn5, have multiple copies in the mitochondrial genome of C. nitidissima (). Five intron-containing genes (ccmFc, nad2, nad4, nad1, nad5) were found, which contained 15 introns totally. Based on 23 conserved protein-coding genes (atp1, atp4, atp6, atp8, atp9, ccmB, ccmC, cox1, cox3, cytb, matR, mttB, nad1, nad2, nad3, nad4, nad5, nad6, nad7, rpl2, rps10, rps19, rps3) in the mitochondrial genomes, the phylogenetic analysis showed that 24 plants were divided into five branches. The following sequences with GenBank accession were used: Glycine max NC_020455 (Chang et al. Citation2013), Glycine soja NC_039768 (Asaf et al. Citation2018), Phaseolus vulgaris NC_045135 (Bi et al. Citation2020), Lotus japonicas NC_016743 (Kazakoff et al. Citation2012), Medicago truncatula NC_029641 (Bi et al. Citation2016), Cucumis sativus NC_016005, Cucurbita pepo NC_014050 (Alverson et al. Citation2010), Citrullus lanatus NC_014043 (Alverson et al. Citation2010), Malus domestica NC_018554 (Goremykin et al. Citation2012), Brassica napus NC_008285 (Handa Citation2003), Brassica oleracea NC_016118 (Chang et al. Citation2011), Raphanus sativus KJ716484 (Jeong et al. Citation2016), Arabidopsis thaliana NC_001284 (Giegé and Brennicke Citation1999), Gossypium barbadense NC_028254 (Tang et al. Citation2015), Gossypium raimondii NC_029998 (Bi et al. Citation2016), Asclepias syriaca NC_022796 (Straub et al. Citation2013), Rhazya stricta NC_024293 (Park et al. Citation2014), Nicotiana attenuate NC_036467, Solanum lycopersicum NC_035963 (Xu et al. Citation2017), Helianthus annuus NC_023337 (Bock et al. Citation2014), Camellia sinensis NC_043914 (Rawal et al. Citation2020), Camellia nitidissima ON645224 (unpublished), Vitis vinifera NC_012119 (Goremykin et al. Citation2009), Ginkgo biloba NC_027976 (Guo et al. Citation2016). The Ginkgo biloba (NC_027976) used as outgroup was divided into one branch; the grape was divided into one branch alone; C. nitidissima and six plants were divided into one branch; two plants of Malvaceae and four plants of Cruciferous were divided into another branch and the remaining nine plants were divided into final branch (). In the phylogenetic tree, C. nitidissima was clustered into a branch with the plants of Theaceae, Apocynaceae, Solanaceae and Compositae, which had high bootstrap value and fited to the angiosperm phylogeny group classification (APG IV). Moreover, C. nitidissima was closely related to Camellia sinensis, which all belonged to Camellia genus of the family Theaceae, with 100 bootstrap values. Therefore, it is thought that the phylogenetic analysis in this study can distinguish species of plant by using sequences of mitochondrial gene.
Figure 2. The gene map of the complete mitochondrial genome of C. nitidissima. Genes indicated in the inner circle are transcribed clockwise, and those indicated in the outer circle are transcribed counterclockwise. Different functional groups of genes are color coded. The darker gray corresponds to DNA G + C content, while the lighter gray corresponds to A + T content.
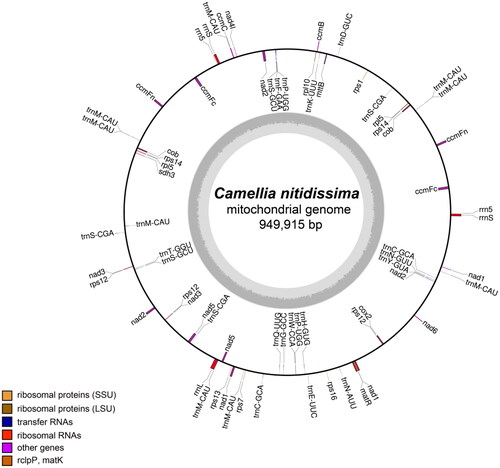
Figure 3. Used the Ginkgo biloba (NC_027976) as outgroup, the phylogenetic tree of 24 plants was constructed with maximum-likelihood method by PhyloSuite. The numbers above the branches were the bootstrap value. The 23 conserved protein-coding genes (atp1, atp4, atp6, atp8, atp9, ccmB, ccmC, cox1, cox3, cytb, matR, mttB, nad1, nad2, nad3, nad4, nad5, nad6, nad7, rpl2, rps10, rps19, rps3) in the mitochondrial genomes were used to construct the phylogenetic.
Discussion and conclusion
The event of horizontal gene transfer can be identified by comparing mitochondrial genome of C. nitidissima with the chloroplast genome in future studies, which will provide a basis for the functional study of horizontal gene transfer sequences. This complete mitochondrial genome of C. nitidissima will benefit the evolution study, germplasm identification and development of molecular markers.
Ethical approval
The data collection of plants was carried out with the permission of Yulin Normal University and complied with local (Yulin, Guangxin, China) legislation. The research involved Camellia nitidissima, which was an endangered species, so in this study we complied with the policies of the International Union for Conservation of Nature (IUCN), the Convention on Biological Diversity and the Convention on the Trade in Endangered Species of Wild Fauna and Flora, and tried ours best to protect the resources of C. nitidissima.
Author contribution
Q. L. and H. L. were involved in the conception and design; B. L. and Y. Z. contributed the sample collection; B. L. and Q. L. performed the analysis and interpretation of the data; Q. L., X. Z. and H. L. contributed the drafting of the paper; Y. C. and S. X. revised it critically for intellectual content. All authors were involved in the final approval of the version to be published. All authors agree to be accountable for all aspects of the work.
Supplemental Material
Download MS Word (31.4 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at [https://www.ncbi.nlm.nih.gov] under the accession no. ON645224.1. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA899601, SRR22240446, SRR22240447, and SAMN31665678, respectively.
Additional information
Funding
References
- Alverson AJ, Wei XX, Rice DW, Stern DB, Barry K, Palmer JD. 2010. Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol Biol Evol. 27(6):1436–1448.
- Asaf S, Khan AL, Al-Harrasi A, Kim TH, Lee IJ. 2018. The first complete mitochondrial genome of wild soybean (Glycine soja). Mitochondrial DNA B Resour. 3(2):527–528.
- Bi CW, Lu NL, Xu YQ, He CP, Lu ZH. 2020. Characterization and analysis of the mitochondrial genome of common bean (Phaseolus vulgaris) by comparative genomic approaches. IJMS. 21(11):3778.
- Bi CW, Paterson AH, Wang XL, Xu YQ, Wu DY, Qu YS, Jiang A, Ye QL, Ye N. 2016. Analysis of the complete mitochondrial genome sequence of the diploid cotton Gossypium raimondii by comparative genomics approaches. Biomed Res Int. 2016:5040598.
- Bi CW, Wang XL, Xu YQ, Wei SY, Shi Y, Dai XG, Yin TM, Ye N. 2016. The complete mitochondrial genome of Medicago truncatula. Mitochondrial DNA B Resour. 1(1):122–123.
- Bock DG, Kane NC, Ebert DP, Rieseberg LH. 2014. Genome skimming reveals the origin of the Jerusalem Artichoke tuber crop species: neither from Jerusalem nor an artichoke. New Phytol. 201(3):1021–1030.
- Chang SX, Wang YK, Lu JJ, Gai JY, Li JJ, Chu P, Guan RZ, Zhao TJ. 2013. The mitochondrial genome of soybean reveals complex genome structures and gene evolution at intercellular and phylogenetic levels. PLoS One. 8(2):e56502.
- Chang SX, Yang TT, Du TQ, Huang YJ, Chen JM, Yan JY, He JB, Guan RZ. 2011. Mitochondrial genome sequencing helps show the evolutionary mechanism of mitochondrial genome formation in Brassica. BMC Genomics. 12:497.
- Giegé P, Brennicke A. 1999. RNA editing in Arabidopsis mitochondria effects 441 C to U changes in ORFs. Proc Natl Acad Sci USA. 96(26):15324–15329.
- Goremykin VV, Lockhart PJ, Viola R, Velasco R. 2012. The mitochondrial genome of Malus domestica and the import-driven hypothesis of mitochondrial genome expansion in seed plants. Plant J. 71(4):615–626.
- Goremykin VV, Salamini F, Velasco R, Viola R. 2009. Mitochondrial DNA of Vitis vinifera and the issue of rampant horizontal gene transfer. Mol Biol Evol. 26(1):99–110.
- Guo WH, Grewe F, Fan WS, Young GJ, Knoop V, Palmer JD, Mower JP. 2016. Ginkgo and Welwitschia mitogenomes reveal extreme contrasts in gymnosperm mitochondrial evolution. Mol Biol Evol. 33(6):1448–1460.
- Handa H. 2003. The complete nucleotide sequence and RNA editing content of the mitochondrial genome of rapeseed (Brassica napus L.): comparative analysis of the mitochondrial genomes of rapeseed and Arabidopsis thaliana. Nucleic Acids Res. 31(20):5907–5916.
- He D, Li X, Sai X, Wang L, Li S, Xu Y. 2018. Camellia nitidissima C.W. Chi: a review of botany, chemistry, and pharmacology. Phytochem Rev. 17(2):327–349.
- Hou X, Du H, Yang R, Qi J, Huang Y, Feng S, Wu Y, Lin S, Liu Z, Jia AQ, et al. 2018. The antitumor activity screening of chemical constituents from Camellia nitidissima Chi. Int J Mol Med. 41(5):2793–2801.
- Jeong YM, Chung WH, Choi AY, Mun JH, Kim N, Yu HJ. 2016. The complete mitochondrial genome of cultivated radish WK10039 (Raphanus sativus L.). Mitochondrial DNA A DNA Mapp Seq Anal. 27(2):941–942.
- Jiang L, Fan Z, Tong R, Zhou X, Li J, Yin H. 2020. Functional diversification of the dihydroflavonol 4-reductase from Camellia nitidissima Chi. in the control of polyphenol biosynthesis. Genes. 11(11):1341.
- Jin JJ, Yu WB, Yang JB, Song Y, dePamphilis CW, Yi TS, Li DZ. 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):241.
- Kazakoff SH, Imelfort M, Edwards D, Koehorst J, Biswas B, Batley J, Scott PT, Gresshoff PM. 2012. Capturing the biofuel wellhead and powerhouse: the chloroplast and mitochondrial genomes of the leguminous feedstock tree Pongamia pinnata. PLoS One. 7(12):e51687.
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27(5):722–736.
- Li X, Wang J, Sun Z, Wang J, Yin H, Fan Z, Li J. 2019. Flavonoid components and their effects on flower colors in Camellia nitidissima, White C. japonica and their three hybrid cultivars. Acta Horticulturae Sinica. 46(06):1145–1154.
- Lin JN, Lin HY, Yang NS, Li YH, Lee MR, Chuang CH, Ho CT, Kuo SC, Way TD. 2013. Chemical constituents and anticancer activity of yellow camellias against MDA-MB-231 human breast cancer cells. J Agric Food Chem. 61(40):9638–9644.
- Park S, Ruhlman TA, Sabir JS, Mutwakil MH, Baeshen MN, Sabir MJ, Baeshen NA, Jansen RK. 2014. Complete sequences of organelle genomes from the medicinal plant Rhazya stricta (Apocynaceae) and contrasting patterns of mitochondrial genome evolution across asterids. BMC Genomics. 15(1):405.
- Rawal HC, Kumar PM, Bera B, Singh NK, Mondal TK. 2020. Decoding and analysis of organelle genomes of Indian tea (Camellia assamica) for phylogenetic confirmation. Genomics. 112(1):659–668.
- Straub SC, Cronn RC, Edwards C, Fishbein M, Liston A. 2013. Horizontal transfer of DNA from the mitochondrial to the plastid genome and its subsequent evolution in milkweeds (apocynaceae). Genome Biol Evol. 5(10):1872–1885.
- Tang MY, Chen ZW, Grover CE, Wang YM, Li SS, Liu GZ, Ma ZY, Wendel JF, Hua JP. 2015. Rapid evolutionary divergence of Gossypium barbadense and G. hirsutum mitochondrial genomes. BMC Genomics. 16:770.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq-versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45(W1):W6–W11.
- Wang W, Liu H, Wang Z, Qi J, Yuan S, Zhang W, Chen H, Finley JW, Gu L, Jia AQ. 2016. Phytochemicals from Camellia nitidissima Chi inhibited the formation of advanced glycation end-products by scavenging methylglyoxal. Food Chem. 205:204–211.
- Xu YQ, Zhang FQ, Hu K. 2017. The complete mitochondrial genome sequence of an annual wild tobacco Nicotiana attenuata. Mitochondrial DNA B Resour. 2(2):924–925.
- Zhang D, Gao F, Jakovlić I, Zou H, Zhang J, Li WX, Wang GT. 2020. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour. 20(1):348–355.
- Zhou X, Li J, Zhu Y, Ni S, Chen J, Feng X, Zhang Y, Li S, Zhu H, Wen Y. 2017. De novo assembly of the Camellia nitidissima transcriptome reveals key genes of flower pigment biosynthesis. Front Plant Sci. 8:1545.