
Abstract
Begonia ferox C.I Peng & Yan Liu (2013) was rated as endangered according to Red List of Chinese Plants. In this study, we report the complete chloroplast genome of B. ferox. The chloroplast genome is 169,114 bp in length as the circular, with the GC content of 35.5%, composed by a large single-copy (LSC) region of 75,887 bp, a small single-copy (SSC) region of 18,105 bp, and two inverted repeat regions (IRs) of 37,561 bp in each. The genome comprises 174 encoded genes in total, including 114 protein-coding genes, eight ribosomal RNA genes, and 52 transfer RNA genes. Phylogenetic analysis indicated that B. ferox is genetically closest to B. gulongshanensis.
Introduction
Begonia (Begoniaceae) is one of the most diverse plant groups and the fifth or sixth largest angiosperm genus in the world, composed of more than 2000 recognized species names. In the past 20 years, the number of species of the genus has increased dramatically, rising from 80 to 200 species in China (Tian et al. Citation2018). Begonia ferox C.I Peng & Yan Liu (2013) is a perennial herb () of Sect. Coelocentrum in the genus Begonia (Begoniaceae) (Peng et al. Citation2013). Begonia ferox is mainly distributed on limestone rocks with ample fallen leaves or on exposed rock slopes in evergreen broad-leaved forests in Guangxi Zhuangzu Autonomous Region, Longzhou County only, very rare. The leaf of B. ferox covered with rounded swellings like blisters, and intercostal area densely distributed with dark brown and hair-tipped bubbles. Thus, this species has high ornamental value. Due to narrow habitat and commercial collection, it has been facing serious threat. According to the latest red list of Chinese plants, B. ferox is assessed as endangered. Therefore, we report the chloroplast genome sequence of B. ferox to provide a basis for further study better of this species and deeply exploration of the phylogenetic history of Begoniaceae.
Figure 1. Begonia ferox C.-I Peng & Yan Liu. (A) Cultivated plant at fruiting stage; (B) leaf adaxial surface with humps; (C) leaf back surface. All of them were photographed by Zhou in the greenhouse of Guangxi Institute of Botany.

Materials and methods
The fresh leaves of B. ferox were collected from the greenhouse (110°18′17.80″E, 25°04′49.65″N) in Guangxi Institute of Botany, Chinese Academy of Sciences. B. ferox sampling was permitted by the Guangxi Science and Technology Base and Special Talents Program, China (Guike AD20325009). The corresponding specimen was deposited at Herbarium of Guangxi Institute of Botany (http://ibk.gxib.cn/, contact person: Yusong Huang, and email: [email protected]) under voucher number IBK00445109. Total genomic DNA in leaf tissue samples was extracted using CTAB method following the manufacturers’ protocols (Doyle Citation1987), was assessed using Agilent 5400 Tapestation (Agilent Technology Ltd., Santa Clara, CA) for quality control (Figure S1). Then, paired-end sequenced by Illumina HiSeq Xten platform (Illumina, San Diego, CA). A total of 18,413,794 clean reads (% reads with ≥ Q30 score = 93.00%) (Figure S2) were generated, then they were assembled by SPAdes 3.11.0 (Bankevich et al. Citation2012). To check the accuracy of assembly, we used BWA software to compare the Illumina short sequence with the chloroplast sequence, and used SAMtools (Li et al. Citation2009) depth to calculate the coverage (Figure S3). Finally, the assembled genome was annotated using PGA software (Qu et al. Citation2019) and further cp genome map was done using OGDRAW (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html) (Greiner et al. Citation2019), with Begonia guangxiensis (GenBank accession no. NC_046385) as the reference genome.
Results
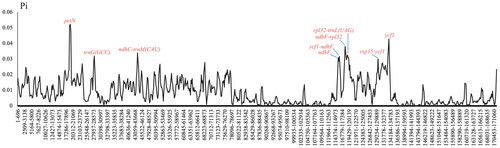
The chloroplast genome of B. ferox (GenBank accession no. NC_067030.1) is an enclosed loop molecule with a length of 169,114 bp (). The total GC content is 35.5%. The genome contains a large single-copy (LSC) region of 75,887 bp, a small single-copy (SSC) region of 18,105 bp, and a pair of inverted repeat regions (IRA and IRB) of 37,561 bp. PGA annotation results showed that chloroplast genes contained 144 genes (119 unique genes), including 94 protein-coding genes (80 unique genes), eight rRNAs (four unique genes) and 42 tRNA genes (35 unique genes). Every 28 genes were found to be duplicated in the IR regions, including 12 PCGs genes (psbI, psbK, rps16, psbA, rpl2, rpl23, ycf2, ycf15, ndhB, rps7, rps12, and ycf1), 12 tRNA genes (trnN-GUU, trnR-ACG, trnA-UGC, trnI-GAU, trnV-GAC, trnL-CAA, trnI-CAU, trnH-GUG, trnK-UUU, trnQ-UUG, trnS-GCU, and trnG-UCC), and four rRNA genes (rRNA4.5, rRNA5, rRNA16, and rRNA23). The introns were contained in seven protein-coding genes and six tRNA genes, while ycf3 and clpP contain two introns. In particular, a trans-spliced gene rps12 was detected by CPGview (http://www.1kmpg.cn/cpgview/) (Figure S4). In addition, the cp genomes of four Begonia species in China (B. ferox, B. gulongshanensis, B. coptidifolia, and B. pulchrifolia) were aligned and compared to screen (used DnaSP) single-nucleotide polymorphisms (SNPs) with higher nucleotide diversity (Ning et al. 2020). As a result, the nucleotide variability (Pi) values were ranged from 0.008 to 0.052 (). We found nine of these variable regions (Pi > 0.03) including petN, trnG(GCC), ndhC-trnM(CAU), ndhF, ycfl-ndhF, ndhF-rpl32, rpl32-trnL(UAG), rsp15-ycf1, and ycf1 showed high level of intrageneric variation.
Figure 2. Complete chloroplast (cp) genome map of B. ferox. Color coding of genes is based on functional groups they belong to. Dark gray color of inner circle indicates GC content.
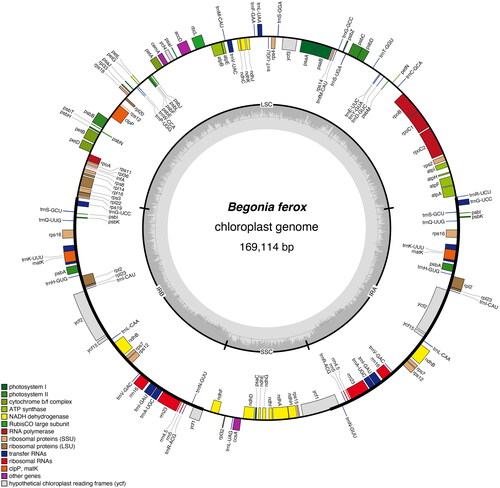
Figure 3. Result of comparative analysis of the nucleotide variability (Pi) values among four Begonia species in China.
Discussion and conclusions
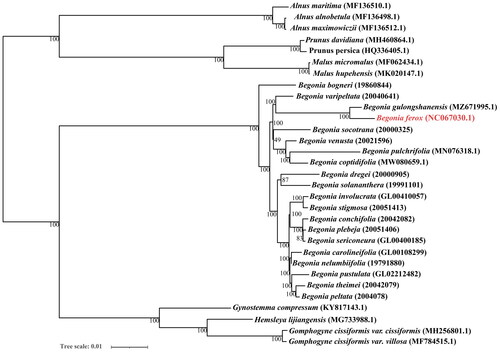
To further analyze the phylogenetic relationship of B. ferox, all 31 sequences (14 downloaded from NCBI and 16 downloaded from Dryad (Harrison et al. Citation2016)) were aligned via mafft v7.505 (Katoh and Standley Citation2013), applying the iterative refinement method (FFT-NS-i) and using default parameter settings (gap opening penalty: 1.53, offset-value: 0.0). Then, the phylogenetic maximum-likelihood (ML) tree was constructed under the GTR + I + G model via W-IQ-TREE (Trifinopoulos et al. Citation2016) and all nodes were inferred from 1000 bootstrap values, finally visually inspected and manually adjusted in the iTOL (Letunic and Bork Citation2007). The phylogenetic analysis indicated that B. ferox is closely related to B. gulongshanensis (). We speculated that B. ferox and B. gulongshanensis both belong to the Sect. Coelocentrum, and their type locality is very close, located in the west of Guangxi Zhuang Autonomous Region.
Figure 4. The maximum-likelihood (ML) phylogenetic tree based on the complete chloroplast genomes of B. ferox and 30 other published species. Bootstrap support values (1000 repetitions) are shown next to the nodes.
Author contributions
Tao Deng: contributed to the conception of the study; Qiongyue Liang: prepared the sample and acquired the data; Xuan Yang: contributed significantly to analysis and manuscript preparation; Yangming Zhou: performed the experiment, analyzed the data, and wrote the manuscript; Xinghua Hu: helped perform the analysis with constructive discussions; we agree to be accountable for all aspects of the work.
Ethical approval
This document does not contain any research with human participants or animals by any authors. We strictly comply with the Regulations of the People’s Republic of China on the Protection of Wild Plants, the International Union for Conservation of Nature, the Convention on Biological Diversity and the Convention on International Trade in Endangered Species of Wild Fauna and Flora. Our study approved by Guilin City (Guangxi, China) and Guangxi Institute of Botany of the Chinese Academy of Sciences of Guangxi Zhuang Autonomous Region (Guangxi, China). Therefore, all plant experiments were conducted according to the policies related to endangered species described in these permits and national regulations.
Supplemental Material
Download MS Word (504.6 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The complete sequence of the chloroplast genome of B. ferox has been deposited in GenBank at https://www.ncbi.nlm.nih.gov/, and the accession no. was OP278659. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA872291, SRR21185740, and SAMN30451026, respectively. The data are morally correct and does not violate the protection of human subjects or other valid ethical, privacy, or security concerns.
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477. doi: 10.1089/cmb.2012.0021.
- Doyle JJ. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Greiner S, Lehwark P, Bock R. 2019. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47(W1):W59–W64. doi: 10.1093/nar/gkz238.
- Harrison N, Harrison RJ, Kidner CA. 2016. Comparative analysis of Begonia plastid genomes and their utility for species-level phylogenetics. PLOS One. 11(4):e0153248. doi: 10.1371/journal.pone.0153248.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780. doi: 10.1093/molbev/mst010.
- Letunic I, Bork P. 2007. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 23(1):127–128. doi: 10.1093/bioinformatics/btl529.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25(16):2078–2079. doi: 10.1093/bioinformatics/btp352.
- Ning C, Liao B-S, Cong-Lian L, Shi-Feng LI, Hao Z, Jiang XU, Li X-W, Shi-Lin C. 2020. Complete chloroplast genome of Salvia plebeia: organization, specific barcode and phylogenetic analysis. Chin J Nat Med. 18(8):563–572.
- Peng CI, Yang HA, Kono Y, Chung KF, Huang YS, Wu WH, Liu Y. 2013. Novelties in Begonia sect. Coelocentrum: B. longgangensis and B. ferox from limestone areas in Guangxi, China. Bot Stud. 54(1):44. doi: 10.1186/1999-3110-54-44.
- Qu XJ, Moore MJ, Li DZ, Yi TS. 2019. PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods. 15(1):1–12. doi: 10.1186/s13007-019-0435-7.
- Tian D, Xiao Y, Tong Y, Fu N, Liu Q, Li C. 2018. Diversity and conservation of Chinese wild begonias. Plant Divers. 40(3):75–90. doi: 10.1016/j.pld.2018.06.002.
- Trifinopoulos J, Nguyen LT, von Haeseler A, Minh BQ. 2016. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44(W1):W232–W235. doi: 10.1093/nar/gkw256.