
Abstract
Jacobaea maritima is an important horticulture plant in the genus Jacobaea. Here, we assembled the complete chloroplast genome of J. maritima. The chloroplast genome was 153,857 bp in length, with a pair of inverted repeat regions (IRs) (27,936 bp) separated by a large single-copy region (LSC) (82,771 bp) and a small single-copy region (SSC) (15,214 bp). The complete chloroplast genome contained 112 unique genes, including 79 protein-coding genes, 29 tRNA genes, and 4 rRNA genes. The phylogenetic analysis showed Jacobaea was more closely related to Senecio, Crassocephalum and Gynura. The chloroplast genome of J. maritima can provide data to support future phylogenetic studies of Jacobaea.
Background
Jacobaea maritima (L.) Pelser & Meijden belongs to the tribe Senecioneae branch in the family Asteraceae, and prefers to grow in cliffs and rocky coastal areas, usually distributed in the Mediterranean region, northwest Africa, southern Europe and western Asia, while in China it is mainly used as an ornamental plant (Galasso and Bartolucci Citation2015; Passalacqua et al. Citation2008). The Jacobaea was rich in secondary metabolites, including flavonoids, pyrrolizidine alkaloids, triterpenoids and aromatic acids (Maggio et al. Citation2015). Therefore, Jacobae species had become important for its ornamental and medicinal value and were grown in most areas with great climatic adaptability to different regions.
The phylogenetic study of Jacobaea is mainly based on plastid fragments and nuclear genes (Figure S1). In 2002, the molecular phylogeny of Jacobaea was studied to clarify species composition and interspecific relationships of Jacobaea (Pelser et al. Citation2002). A resolution of 60 representative species of the tribe Senecioneae based on DNA sequence data from the plastid sequences (trnT-trnL, trnL intron, trnK intron, and matK gene) and nuclear sequences (ITS1, 5.8S, and ITS2) revealed that Jacobaea was a strongly supported monophyletic group. The genera Emilia, Packera and Pseudogynoxys formed the sister clade of Jacobaea, but this relationship lacked strong bootstrap support. In 2003, the species composition and molecular phylogeny of Jacobaea were studied to identify the closest relatives of J. vulgaris (Pelser et al. Citation2003). Maximum parsimony and Bayesian inference analyses of DNA sequence data of the plastid (trnT-L, trnL intron, trnK intron, and psbA-trnH) and nuclear genome (ITS1, 5.8S, and ITS2) showed these markers to be suitable to assess the species composition of Jacobaea. In 2008, researchers performed phylogenetic analysis of the J. maritima group based on ITS1 sequence (Passalacqua et al. Citation2008). In 2021, a systematic study of diversity, polyploidy and morphology in the family Asteraceae based on single-copy nuclear genes, showed that the tribe Senecioneae was a monophyletic group and Jacobaea had a close relationship with Senecio and Emilia (Zhang et al. Citation2021).
However, studies on the chloroplast genome of J. maritima are still lacking, and phylogenetic questions regarding Jacobaea have not been addressed. In this study, we sequenced and assembled the chloroplast genome of J. maritima and performed comparative genomic analyses with the intention of further revealing the chloroplast genome characteristics of Jacobaea species. These results will provide a data base for phylogenetic studies of the tribe Senecioneae and Jacobaea species.
Materials and methods
Plant material, DNA extraction and sequencing
Fresh leaves of J. maritima were sampled from second-year old plants in Xinyang, Henan, China (; 114.21667° E, 32.28333° N). The voucher specimen was deposited in the herbarium of Xinyang Agriculture and Forestry University (voucher number: XJM22004; Mr. Gong, [email protected]). The total genomic DNA was extracted using the HiPure Plant DNA Mini Kit (Magen, Guangzhou, China). A DNA library with an average length of 500 bp was constructed according to the instructions of the TruSeq DNA PCR-Free LT Sample Prep kit, followed by high-throughput sequencing (paired-end 250 bp) using the Illumina NovaSeq6000 platform. Raw reads were filtered using Trimmomatic v. 0.39 with default parameters retrieving the clean data.
Figure 1. Reference images of J. maritima (in this study, this picture was taken by Kai Zhang). the voucher specimen was deposited in the herbarium of Xinyang Agriculture and Forestry University (voucher number: XJM22004; 114.21667° E, 32.28333° N).

Genome assembly and annotation
De novo genome assembly from the clean data was accomplished using NOVOPlasty software (Dierckxsens et al. Citation2017), with the seed input file consisting of the protein-coding genes from the reference genome (J. vulgaris, GenBank: NC_015543) with k-mer setting of 39. Then we used samtools v1.7 (Li et al. Citation2009) and bedtools v2.28 (Quinlan and Hall Citation2010) for coverage depth detection, setting the window and step size to 200 bp, respectively. We also used MUMmer v4.0 (Marçais et al. Citation2018) for collinearity analysis. The chloroplast genome was annotated by using CPGAVAS2 (Shi et al. Citation2019), PGA (Qu et al. Citation2019) and Geneious Prime v. 2022.2.2 with a reference genome (J. vulgaris, GenBank: NC_015543). GB2sequin (https://chlorobox.mpimp-golm.mpg.de/GenBank2Sequin.html) was used to confirm the annotation result, and then CPGView (Liu et al. Citation2023) (http://www.1kmpg.cn/cpgview/) was used to test the accuracy of these genes. The genome map was drawn by Chloroplot (https://irscope.shinyapps.io/Chloroplot/).
Repeat sequence and IR boundary analysis
The simple sequence repeats (SSRs) were identified using the online website MISA (https://webblast.ipk-gatersleben.de/misa/), including mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides with minimum numbers of 10, 5, 4, 3, 3, and 3, respectively (Beier et al. Citation2017). Additionally, REPuter (https://bibiserv.cebitec.uni-bielefeld.de/reputer/) was used to calculate palindromic repeats, forward repeats, reverse repeats, and complementary repeats with the following settings: minimal repeat size of 30 bp (Kurtz et al. Citation2001). Furthermore, comparisons between the borders of the IR, SSC, and LSC regions were generated using IRscope (Amiryousefi et al. Citation2018).
Phylogenetic analysis
To confirm the phylogenetic position of J. maritima, the chloroplast genomes of 20 Asteraceae species and one Brassicaceae species were downloaded from GenBank. The Arabidopsis thaliana was used as outgroup. We extracted 75 common protein-coding genes from the genome annotation files using PhyloSuite v. 1.2.2 (Zhang et al. Citation2020). Each protein-coding gene sequence was aligned using MAFFT v. 7.4 (Katoh and Standley Citation2013), and the 75 aligned sequences were concatenated. Based on the matrix of concatenate sequence, a phylogenetic tree was constructed using the maximum likelihood (ML) method implemented in IQ-TREE v. 2.1.2 (Nguyen et al. Citation2015), and the best model was inferred from ModleFinder (Kalyaanamoorthy et al. Citation2017). The bootstrap analysis was performed with 1000 replicates. Tree visualization was achieved in Figtree v. 1.4.3 (https://github.com/rambaut/figtree/releases).
Results
General features of the chloroplast genome
We examined the chloroplast genome for completeness, coverage depth and collinearity, and the results showed that the chloroplast genome was reliable (Figure S2). Analysis of the genome annotation results also showed that the cis- and trans-splicing gene annotations were correct (Figure S3). The complete chloroplast genome of J. maritima was a typical circular tetrameric structure of 153,857 bp in length (; ; GenBank accession number: OL960706), consisting of a large single copy (LSC) region (82,771 bp), a small single copy (SSC) region (15,214 bp) and a pair of inverted repeats (IR) (27,936 bp). Percentages of the four base types for the whole chloroplast genome of J. maritima were 31.43% A, 31.43% T, 18.83% G, and 18.30% C. The chloroplast genome encoded 112 unique genes, including 79 protein-coding genes, 4 ribosomal RNA genes and 29 transfer RNA genes (Table S1). The chloroplast genome had a total GC content of 37.13% and the IR region had a GC content (41.37%) was significantly higher than that of the LSC region (35.43%) and the SSC region (31.81%). In the study, we detected 128 SSRs in the chloroplast genome of J. maritima, including 34 mononucleotides, 41 dinucleotides, 19 trinucleotides, 24 tetranucleotides, 7 pentanucleotides and 3 hexanucleotides (). Most SSRs were mononucleotides and dinucleotides, accounting for 58.59% of the total. In the chloroplast genomes of J. maritima and J. vulgaris, SSRs were most abundant in the LSC region and least in the IR region, and mainly concentrated in the coding region, with fewer SSRs in the non-coding region (; Table S2). This phenomenon may be related to the sequence conservativeness and GC content of different regions, etc. In addition, we detected 4 interspersed repeats in the chloroplast genome of J. maritima, including one forward repeat and 3 palindromic repeats.
Figure 2. The chloroplast genome map of J. maritima. Genes on the inside of the circle are transcribed in a clockwise direction and genes on the outside of the circle are transcribed in a counter-clockwise direction.
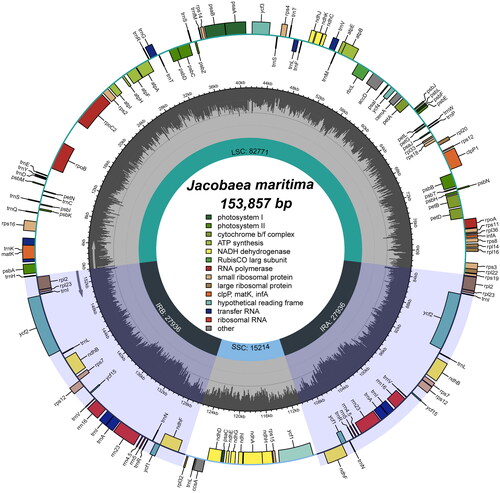
Table 1. Summary of the chloroplast genomes of two Jacobaea species.
IR boundaries analysis
In general, the length variation of the LSC/SSC region was lower than that of the IRa/IRb region. The chloroplast genome of J. maritima showed the expansion of the IR region and contraction of the SSC region compared to the chloroplast genome of J. vulgaris (Figure S4). In the chloroplast genome of J. maritima, rps15 and rpl32 were located within the SSC/IR boundary, and ndhF and rpl2 were located outside the SSC/IR boundary. In contrast, in the chloroplast genome of J. vulgaris, ycf1 was located within the SSC/IR boundary, ndhF was located within the SSC/IR boundary, and rpl2 and trnN were located outside the SSC/IR boundary.
Phylogenetic analysis
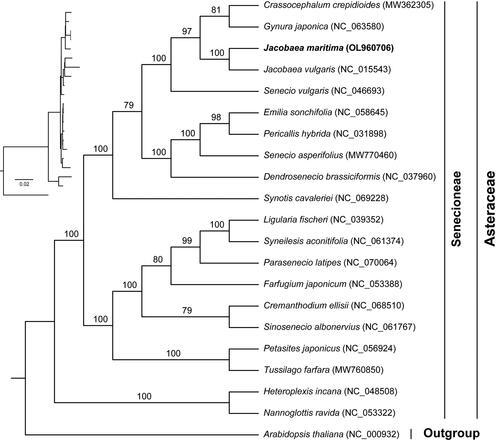
To understand the phylogenetic position of J. maritima in the Asteraceae, we performed a phylogenetic analysis. The maximum-likelihood tree was constructed using IQ-TREE v. 2.1.2 (Nguyen et al. Citation2015) with the best-fit model of TVM + F+R4. The phylogenetic analysis showed high bootstrap for most of nodes in the phylogenetic tree, showing the reliability of the phylogeny (). Our results clearly showed that Jacobaea was more closely related to Senecio, Crassocephalum and Gynura.
Figure 3. Phylogenetic tree based on the concatenated sequences of 75 common protein-coding genes in 21 species using maximum-likelihood (ML) method. The value above the branch indicates the bootstrap value.
Discussion
Compared with nuclear and mitochondrial genomes, chloroplast genomes are highly conserved and have been widely used in phylogenetic and evolutionary studies. With the development of high-throughput sequencing technology, chloroplast genome sequences play an important role in phylogenetic study (Li et al. Citation2021). In this study, the chloroplast protein-coding genes tree showed high support with credibility. Our phylogenetic results were also in general agreement with the previous results (Figure S1) (Passalacqua et al. Citation2008; Zhang et al. Citation2021), which showed that Jacobaea was more closely related to Senecio, Crassocephalum and Gynura. Future enrichment of chloroplast genomic data of Jacobaea species is needed to further resolve intra-generic phylogenetic relationships. However, these results are limited due to the matrilineal inheritance of the chloroplast genome (Krawczyk et al. Citation2018). An accurate phylogenetic relationship still requires an integrated analysis of the nuclear and organelle genomes (Górniak et al. Citation2010). More molecular data is needed in the future to determine the relationships between Jacobaea and other genus in the family Asteraceae.
Conclusion
In this study, the chloroplast genome of J. maritima was de novo assembled with short reads. This chloroplast genome had a typical tetrameric structure similar to that of most angiosperms. In addition, the phylogenetic tree strongly supported the phylogenetic position of J. maritima, showing that Jacobaea was more closely related to Senecio, Crassocephalum and Gynura. Thus, the chloroplast genome of J. maritima not only enriches the genomic information of Jacobaea, but also lays the foundation for understanding the evolution of Asteraceae species.
Ethical approval
The author has read the manuscript and has approved this submission. Ethical statement is not applicable. The research on plants used in this study, including the collection of plant material has been carried out in accordance with guidelines provided by our institution. Field studies in our manuscript have complied with local legislation and appropriate permissions/license were granted while taking samples from a preserved/protected land.
Author contributions
Shoufu Gong directed the study and designed the experiments. Kai Zhang and Shoufu Gong performed the data processing. Kai Zhang drafted the manuscript. All authors approved the final draft.
Supplemental Material
Download MS Excel (17.4 KB)Supplemental Material
Download MS Excel (11.8 KB)Supplemental Material
Download JPEG Image (212.6 KB){kind=link}
Supplemental Material
Download JPEG Image (449.8 KB){kind=link}
Supplemental Material
Download JPEG Image (471.8 KB){kind=link}
Supplemental Material
Download JPEG Image (3.8 MB){kind=link}
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The contact person of the specimen is Shoufu Gong ([email protected]). The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at (https://www.ncbi.nlm.nih.gov/) under the accession no. OL960706. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA775801, SRX12813512, and SAMN22625029 respectively.
Additional information
Funding
References
- Amiryousefi A, Hyvönen J, Poczai P. 2018. IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics. 34(17):3030–3031. doi:10.1093/bioinformatics/bty220.
- Beier S, Thiel T, Münch T, Scholz U, Mascher M., 2017. MISA-web: a web server for microsatellite prediction. Bioinformatics. 33(16):2583–2585. doi:10.1093/bioinformatics/btx198.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18. doi:10.1093/nar/gkw955.
- Galasso G, Bartolucci F. 2015. Four new combinations in Jacobaea Mill. (Asteraceae, Senecioneae) for the European flora. Nat Hist Sci. 2(2):95–96. doi:10.4081/nhs.2015.246.
- Górniak M, Paun O, Chase MW. 2010. Phylogenetic relationships within Orchidaceae based on a low-copy nuclear coding gene, Xdh: congruence with organellar and nuclear ribosomal DNA results. Mol Phylogenet Evol. 56(2):784–795. doi:10.1016/j.ympev.2010.03.003.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS., 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589. doi:10.1038/nmeth.4285.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780. doi:10.1093/molbev/mst010.
- Krawczyk K, Nobis M, Myszczyński K, Klichowska E, Sawicki J., 2018. Plastid super-barcodes as a tool for species discrimination in feather grasses (Poaceae: stipa). Sci Rep. 8(1):1924. doi:10.1038/s41598-018-20399-w.
- Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R., 2001. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29(22):4633–4642. doi:10.1093/nar/29.22.4633.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R., 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25(16):2078–2079. doi:10.1093/bioinformatics/btp352.
- Li J, Tang J, Zeng S, Han F, Yuan J, Yu J., 2021. Comparative plastid genomics of four Pilea (Urticaceae) species: insight into interspecific plastid genome diversity in Pilea. BMC Plant Biol. 21(1):25. doi:10.1186/s12870-020-02793-7.
- Liu S, Ni Y, Li J, Zhang X, Yang H, Chen H, Liu C., 2023. CPGView: a package for visualizing detailed chloroplast genome structures. Mol Ecol Resour. 23(3):694–704. doi:10.1111/1755-0998.13729.
- Maggio A, Venditti A, Senatore F, Bruno M, Formisano C., 2015. Chemical composition of the essential oil of Jacobaea maritima (L.) Pelser & Meijden and Jacobaea maritima subsp. bicolor (Willd.) B. Nord. & Greuter (Asteraceae) collected wild in Croatia and Sicily, respectively. Nat Prod Res. 29(9):857–863. doi:10.1080/14786419.2014.991928.
- Marçais G, Delcher AL, Phillippy AM, Coston R, Salzberg SL, Zimin A., 2018. MUMmer4: a fast and versatile genome alignment system. PLoS Comput Biol. 14(1):e1005944. doi:10.1371/journal.pcbi.1005944.
- Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274. doi:10.1093/molbev/msu300.
- Passalacqua NG, Peruzzi L, Pellegrino G. 2008. A biosystematic study of the Jacobaea maritima group (Asteraceae, Senecioneae) in the Central Mediterranean area. Taxon. 57(3):893–906. doi:10.1002/tax.573018.
- Pelser PB, Gravendeel B, van der Meijden R. 2002. Tackling speciose genera: species composition and phylogenetic position of Senecio sect. Jacobaea (Asteraceae) based onplastid and nrDNA sequences. Am J Bot. 89(6):929–939. doi:10.3732/ajb.89.6.929.
- Pelser PB, Gravendeel B, van der Meijden R. 2003. Phylogeny reconstruction in the gap between too little and too much divergence: the closest relatives of Senecio jacobaea (Asteraceae) according to DNA sequences and AFLPs. Mol Phylogenet Evol. 29(3):613–628. doi:10.1016/s1055-7903(03)00139-8.
- Qu X-J, Moore MJ, Li D-Z, Yi T-S., 2019. PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods. 15(1):50. doi:10.1186/s13007-019-0435-7.
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26(6):841–842. doi:10.1093/bioinformatics/btq033.
- Shi L, Chen H, Jiang M, Wang L, Wu X, Huang L, Liu C., 2019. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47(W1):W65–W73. doi:10.1093/nar/gkz345.
- Zhang C, Huang C-H, Liu M, Hu Y, Panero JL, Luebert F, Gao T, Ma H., 2021. Phylotranscriptomic insights into Asteraceae diversity, polyploidy, and morphological innovation. J Integr Plant Biol. 63(7):1273–1293. doi:10.1111/jipb.13078.
- Zhang D, Gao F, Jakovlić I, Zou H, Zhang J, Li WX, Wang GT., 2020. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour. 20(1):348–355. doi:10.1111/1755-0998.13096.