
Abstract
In this study, we assembled the complete chloroplast (cp) genome of Malvaviscus penduliflorus using high-throughput Illumina sequencing reads. The resulting plastome assembly displayed a typical quadripartite structure with a total length of 160,332 bp, containing a pair of inverted repeat regions (IRs) of 26,313 bp separated by a large single-copy region (LSC) of 88,750 bp and a small single-copy region (SSC) of 18,956 bp. The M. penduliflorus cp genome contained 128 genes, and its overall GC content was 36.96%. Phylogenetic analysis among M. penduliflorus and five other Malvaceae species demonstrated that M. penduliflorus was closely related to Urena procumbens and Hibiscus cannabinus. The M. penduliflorus cp genome presented in this study will lay a good foundation for further genetic and genomic studies of the genus Malvaviscus as well as Malvaceae.
1. Introduction
Malvaviscus penduliflorus Moc. & Sessé ex DC. 1824 is an evergreen shrub with 30–60 cm, belonging to the Malvaceae family. It grows well in the environment of high temperature, humidity, or sufficient sunlight, which has good heat resistance, barren resistance, and drought resistance. It is widely distributed in tropical regions of the world, such as Cuba, Mexico, and Colombia. In the field of medical treatment, the extracts of M. penduliflorus have been used to treat high blood pressure, liver diseases, fever prevention, and so on (Delange et al. Citation2012).
The chloroplast genome of angiosperms provides a powerful tool for species tree estimation, population genetic analysis, chloroplast gene function and chloroplast metabolism analysis, and investigation of species adaptation to extreme environments (Mehmood et al. Citation2020a, Citation2020b, Citation2020c). However, the complete chloroplast (cp) genome of M. penduliflorus has not been reported to date, which hampers a deeper understanding of its genomic characterization and evolutionary history.
In this study, we first assembled the cp genome of M. penduliflorus and confirmed its phylogenetic position in Malvaceae. This presented cp genome will provide valuable genomic resource for comparative genomic and genetic studies of the genus Malvaviscus as well as Malvaceae.
2. Materials
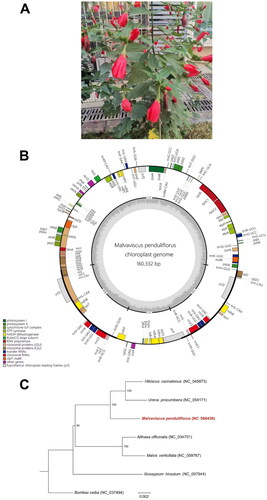
Fresh and mature leaves from an adult plant of M. penduliflorus were taken from Chenghua District, Chengdu, China (30.7000°N, 104.1794°E) (), which were collected according to the Regulations of the People’s Republic of China on the Protection of Wild Plants. A specimen was deposited at the museum of Chengdu University of Traditional Chinese Medicine (Dr. Rong Liu, [email protected]) under the voucher number CDU20201201008.
Figure 1. (A) an Individual plant of M. penduliflorus taken from Chenghua District, Chengdu, China (photoed by Dr. Rong Liu). (B) The detailed genome map of M. penduliflorus cp genome. The large single copy (LSC), small single copy (SSC) region and two inverted repeat regions (IRA and IRB), and GC content (light gray) are shown in the inside track. Gene models including protein-coding genes, tRNA genes, and rRNA genes are shown with various colored boxes in the outer track. (C) Maximum likelihood species tree based on the coding sequences of 75 genes that are presented in all the six cp genomes of Malvaceae species, including M. penduliflorus, U. procumbens (Wang et al. Citation2021), H. cannabinus (Cheng et al. Citation2020), A. officinalis (Zhang et al. Citation2017), M. verticillata (Li et al. Citation2020), and G. hirsutum (Lee et al. Citation2006), using B. ceiba (Gao et al. Citation2018) as outgroup. Bootstrap support values were denoted on each internal node, indicating that M. penduliflorus clustered with U. procumbens and H. cannabinus with 100% bootstrap support.
3. Methods
For whole-genome DNA sequencing, we extracted total genomic DNA using a modified CTAB method (Doyle and Doyle Citation1987). Paired-end Illumina libraries with an insertion size of 350 bp were constructed in accordance with the manufacturer’s instructions and sequenced on an Illumina HiSeq 2500 platform (Illumina, San Diego, CA, USA), resulting in a total of 8.24 Gb Illumina reads. Subsequently, we de novo assembled the cp genome of M. penduliflorus using NOVOPlasty (Dierckxsens et al. Citation2017) using the following parameters: k-mer = 39 and genome range 120,000–200,000, and annotated the plastome assembly using Plann (Huang and Cronk Citation2015) with the Urena procumbens cp genome (GenBank accession number: NC_054171; Wang et al. Citation2021) annotation as the reference. The obvious annotation errors were corrected by Geneious (Kearse et al. Citation2012). We performed phylogentic analysis among M. penduliflorus and five other Malvaceae species, including U. procumbens, Hibiscus cannabinus (NC_045873; Cheng et al. Citation2020), Althaea officinalis (NC_034701; Zhang et al. Citation2017), Malva verticillata (NC_059767; Li et al. Citation2020), and Gossypium hirsutum (NC_007944; Lee et al. Citation2006), using Bombax ceiba (NC_037494; Gao et al. Citation2018) as outgroup. The coding sequences of protein-coding genes present in all the six cp genomes of Malvaceae species were aligned by MAFFT-LINSI v7.313 (Katoh and Standley Citation2013). The resulting alignments were concatenated into a supermatrix, and a maximum likelihood phylogenetic tree was constructed by RAxML v8.2.11 (Stamatakis Citation2006) under the GTRGAMMA model with 500 bootstrap replicates. Further, the cp genomes of M. penduliflorus, U. procumbens, H. cannabinus, A. officinalis, and Malva verticillata were compared using DnaSP v5.10 (Librado and Rozas Citation2009) with the window size and step size of 800 and 200 bp, respectively.
4. Results
Genome sequencing M. penduliflorus resulted in a total of 7.55 Gb Illumina short-read data. The cp genome assembly of M. penduliflorus (GenBank accession number: NC_066439 and ON464180) exhibited a circular structure with a total length of 160,332 bp, containing a pair of inverted repeat regions (IRs) of 26,313 bp separated by a large single-copy region (LSC) of 88,750 bp and a small single-copy region (SSC) of 18,956 bp (). The average coverage depth of M. penduliflorus cp genome was 760× (Supplementary Figure 1). This cp genome contained 138 genes, comprising of 83 protein-coding genes, 37 tRNA genes, and 8 rRNA genes. Among them, 11 protein-coding genes and 8 tRNA genes had a single intron, and three protein-coding genes (rps12, ycf3, and clpP) had two introns. The structure of trans-spliced gene rps12 was verified and shown in Supplementary Figure 2. The overall GC content was 36.96%, and the GC content was 45.76%, 38.18%, and 30.49% for the first, second, and third codons of protein-coding regions, respectively. The 83 protein-coding genes encoded a total of 25,693 codons, and leucine and cystine were the most (10.47%) and least (1.16%) frequently used amino acids. Phylogenetic analysis based on 75 protein-coding genes presented in all the plastomes of M. penduliflorus and five other Malvaceae species showed that M. penduliflorus was closely clustered with U. procumbens and H. cannabinus with 100% bootstrap support (). In addition, comparison of five cp genomes (Supplementary Figure 3) identified several highly variable regions with high nucleotide diversity values (> 0.2), including the genic regions of 7 protein-coding genes (ndhE, ndhG, ndhI, ndhA, ndhH, rps15, and ycf1), 5 tRNA genes (trnN-GUU, trnR-ACG, trnA-UGC, trnI-GAU, and trnV-GAC), and 4 rRNA genes (rrn5, rrn4.5, rrn23, and rrn16). These hotspot regions can be served as potential molecular markers for the phylogenetic analysis of Malvaceae species.
5. Discussion and conclusion
In this study, we first presented the cp genome of M. penduliflorus with a total length of 160,332 bp, a total of 138 genes and an overall GC content of 36.96%. We found that M. penduliflorus was closely clustered with U. procumbens with full support. This genome is the first reported cp genome in the genus Malvaviscus. This cp genome will provide useful genetic resource for further genetic and genomic studies of the genus Malvaviscus and Malvaceae. In the future, increasing number of cp genomes of Malvaceae will provide deeper insights into the evolution of this ecologically and economically important family.
Authors’ contributions
Rong Liu and Dan Wang conceived and supervised the project. Rong Liu and Li Xie analyzed the data and wrote the manuscript. All of the authors have read and approved the final manuscript and have agreed to be accountable for all aspects of the work.
Supplemental Material
Download MS Word (109 KB)Acknowledgments
The authors are really grateful to the open raw genome data form public database.
Disclosure statement
No potential conflict of interest was reported by the author(s). The coauthors do not have any conflict of interest to declare. The authors alone are responsible for the content and composition of the paper. Additionally, fresh and mature leaves from an adult plant of M. penduliflorus were taken with the permission of the community management department.
Data availability statement
The genome sequence data supporting the findings of this study are openly available in GenBank of NCBI (https://www.ncbi.nlm.nih.gov/) under the accession no. NC_066439. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA885282, SRR21748890, and SAMN31083560, respectively.
Additional information
Funding
References
- Cheng Y, Zhang L, Qi J, Zhang L. 2020. Complete chloroplast genome sequence of Hibiscus cannabinus and comparative analysis of the Malvaceae family. Front Genet. 11:227. doi: 10.3389/fgene.2020.00227.
- Delange DM, Rico CLM, Pérez RCS, Canavaciolo VG, Leyes ER. 2012. Determination by GC-MS of the hexane extract components from Malvaviscus penduliflorus flowers growing in Cuba. Anal Chem Lett. 2(3):171–176. doi: 10.1080/22297928.2000.10648266.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18. doi: 10.1093/nar/gkw955.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Gao Y, Wang H, Liu C, Chu H, Yan Y, Tang L. 2018. Complete chloroplast genome sequence of the red silk cotton tree (Bombax ceiba). Mitochondrial DNA B Resour. 3(1):315–316. doi: 10.1080/23802359.2017.1422399.
- Huang DI, Cronk Q. 2015. Plann: a command-line application for annotating plastome sequences. Appl Plant Sci. 3(8):1500026. doi: 10.3732/apps.1500026.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780. doi: 10.1093/molbev/mst010.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649. doi: 10.1093/bioinformatics/bts199.
- Lee SB, Kaittanis C, Jansen RK, Hostetler JB, Tallon LJ, Town CD, Daniell H. 2006. The complete chloroplast genome sequence of Gossypium hirsutum: organization and phylogenetic relationships to other angiosperms. BMC Genomics. 7(1):61. doi: 10.1186/1471-2164-7-61.
- Li R, Liu J, Xu L, Duan B, Qian J. 2020. The complete chloroplast genome of Malva verticillata (Malvaceae). Mitochondrial DNA Part B. 5(2):1609–1610. doi: 10.1080/23802359.2020.1745106.
- Librado P, Rozas J. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 25(11):1451–1452. doi: 10.1093/bioinformatics/btp187.
- Mehmood F, Ubaid Z, Shahzadi I, Ahmed I, Waheed,M. T, Poczai P, Mirza B, Abdullah. 2020a. Plastid genomics of Nicotiana (Solanaceae): insights into molecular evolution, positive selection and the origin of the maternal genome of Aztec tobacco (Nicotiana rustica). PeerJ. 8:e9552. doi: 10.7717/peerj.9552.
- Mehmood F, Ubaid Z, Bao Y, Poczai P, Mirza B, Abdullah. 2020b. Comparative plastomics of Ashwagandha (Withania, Solanaceae) and identification of mutational hotspots for barcoding medicinal plants. Plants. 9(6):752. doi: 10.3390/plants9060752.
- Mehmood F, Shahzadi I, Ahmed I, Waheed M. T, Mirza B, Abdullah. 2020c. Characterization of Withania somnifera chloroplast genome and its comparison with other selected species of Solanaceae. Genomics. 112(2):1522–1530. doi: 10.1016/j.ygeno.2019.08.024.
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 22(21):2688–2690. doi: 10.1093/bioinformatics/btl446.
- Wang JH, Moore MJ, Wang H, Zhu ZX, Wang HF. 2021. Plastome evolution and phylogenetic relationships among Malvaceae subfamilies. Gene. 765:145103. doi: 10.1016/j.gene.2020.145103.
- Zhang N, Ramachandran P, Wen J, Duke JA, Metzman H, McLaughlin W, Ottesen AT, Timme RE, Handy SM. 2017. Development of a reference standard library of chloroplast genome sequences, GenomeTrakrCP. Planta Med. 83(18):1420–1430. doi: 10.1055/s-0043-113449.