
Abstract
In this study, we sequenced the complete mitochondrial genome (mitogenome) of Hypolimnas misippus Linnaeus 1764, which is 15,283 bp in length, containing 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNAs), two ribosomal RNA genes (rRNAs), and an adenine (A) + thymine (T)-rich (D-loop) region. The overall GC level is 19.8%. The phylogenetic position of H. misippus was evaluated using 48 previously published complete mitogenomes, and the results reveals that H. misippus is most closely related to H.bolina.
1. Introduction
The subfamily Nymphalinae (Lepidoptera: Nymphalidae) comprises about 500 species distributed around the world. Several species in the group have been used as model organisms in ecological and evolutionary studies (Brower et al. Citation2000; Boggs et al. Citation2003; Kemp et al. Citation2005). Despite the relatively rich amounts of basic studies about this butterfly group, there is no complete mitochondrial genomes publicly available for Hypolimnas misippus Linnaeus. 1764. Therefore, we sequenced the complete mitochondrial DNA genome of H. misippus to provide baseline data for this species and also to better understand its relationship within the subfamily Nymphalinae.
2. Materials and methods
2.1. Sample collection and preservation
Specimens of female H. misippus () were collected with hand nets from South China Botanical Garden (23°18′35″N, 113°36'56″E), Guangzhou, China. The wings of female butterflies are orange-yellow, and with a small white spot at the top corner. Two rows of small white spots in pairs are arranged on the outer edge of both wings. The color of the back side of the wings is pale, and there are 3 white spots above the middle chamber. There is one black spot on the middle of the anterior margin of the hindwing and one outside the end of the middle wing vein (Chou Citation1998).
Figure 1. Morphological characteristics of Hypolimnas misippus (photographs by JinXu).

2.2. DNA extraction, sequencing, and assembly
Total genomic DNA was extracted from the thorax muscle of a single individual butterfly using the Sangon Animal genome DNA Extraction Kit (Shanghai, China). The voucher specimen was deposited at Southwest Forestry University under the voucher number SCBGJBJD0916 (Jin Xu, [email protected]). After DNA isolation, 1 μg of purified DNA was fragmented and used to construct short-insert libraries (insert size ∼350 bp) according to the manufacturer’s instructions (BIG-500). Then DNA libraries were sequenced by Guangzhou BIO&DATA Biotechnologies Inc. (Guangzhou, China) on the BGI-500 Sequencing System (BGI, Shenzhen, China) using PE 150 bp reads. The filtered sequences were assembled using the SPAdes assembler 3.10.0 (Bankevich et al.Citation2012). The coverage depth (600 × ∼ 4060 ×, mean: 2709.9 ×, Figure S1) was calculated by Geneious R11 (Kearse et al. Citation2012) by mapping the total clean reads to de novo assembled plastome.
2.3. Annotation and analysis
The mito-genome was annotated using MITOS2(Donath et al.Citation2019), BLAST+ (Camacho et al. Citation2009) and tRNAscan (Schattner et al. Citation2005). To investigate its taxonomic relationships, a maximum likelihood (ML) phylogeny was reconstructed based on the whole mitogenome of 46 Nymphalinae butterflies and two outgroup butterflies (Neptis alwina and Parthenos sylvia). All sequences were aligned using the program MAFFT version 7.471 (Katoh and Standley Citation2013), and then a maximum-likelihood (ML) phylogenetic tree was constructed using FastTree version 2.1.10 with Generalized Time-Reversible (GTR) model, statistical support for branches was tested by Shimodaira-Hasegawa (SH) test (Price Citation2010).
3. Results and discussion
3.1. Genomic characterization
The circular mitogenome (Genbank acc. no. OL711673) of H. misippus () is 15,283 bp in length and contains 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNAs), two ribosomal RNA genes (rRNAs), and an adenine (A) + thymine (T)-rich (D-loop) region with 91.0% AT content. Among these, 14 genes are encoded on the L-strand, including four PCGs (ND1, ND4, ND4Land ND5), two rRNA genes, eight tRNA genes (tRNA-Cys, tRNA-Gln, tRNA-His, tRNA-Leu, Trna-Phe, tRNAPro, tRNATyr, and tRNAVal). The remaining 23 genes are encoded on the H strand. The gene composition, order, and direction are similar to the mitogenomes of species from same subfamily of Nymphalidae (Shi et al. Citation2015). The overall AT content (80.2%) is significantly higher than that of GC content (19.8%) in the whole mitogenome.
Figure 2. Mitogenome pattern map of Hypolimnas misippus.
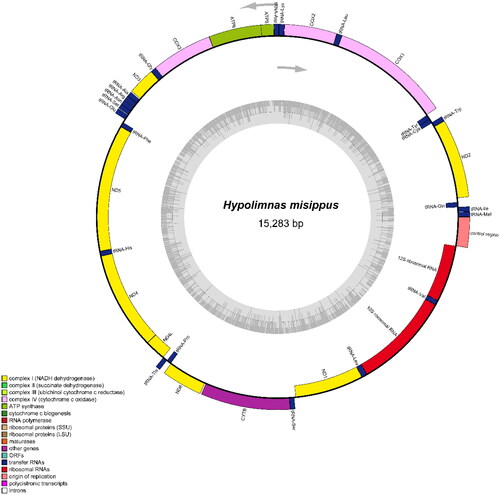
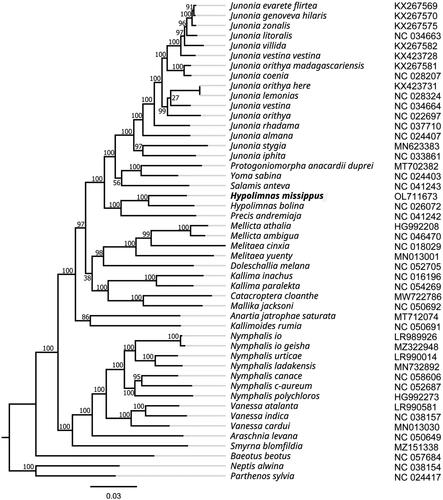
Figure 3. Maximum-likelihood phylogenetic tree based on whole mitogenome from 46 Nymphalinae butterfly and two outgroup butterfly (Neptis alwina and Parthenos sylvia) and the support values are shown at the branches.
3.2. Phylogenetic analysis
The phylogenetic position of H. misippus was inferred by a ML phylogenetic tree using FastTree based on the whole mitogenome of 46 Nymphalinae butterflies and two outgroup butterflies (Neptis alwina and Parthenos sylvia) (Wu et al. Citation2014; Chen et al. Citation2020; Hamilton et al. Citation2020; Liu et al. Citation2020; Li et al. Citation2020; Alexiuk et al. Citation2021; Lalonde Citation2021; Lohse et al. Citation2021; Aguila et al. Citation2021). As shown in , the ML phylogenetic tree shows that H. misippus was most closely related to H. bolina, with support values of 100%. Furthermore, the genus Hypolimnas were clustered together with Precis andremiaja. This relationships is congruent with previous phylogenetic studies (Kim et al. Citation2021).
4. Conclusion
The complete mitochondrial genome of H. misippus was sequenced on the BIG-500 platform to generate a 15,283 bp mitogenome (Genbank accession no. OL711673). The phylogenetic position of H. misippus within the subfamily of Nymphalinae was determined, and the results showed that H. misippus was closely related to H. bolina. Our findings are helpful to understand the phylogenetic status of H. misippus. It also provides baseline molecular data for future studies on the evolutionary relationships within the Nymphalidae.
Ethical approval
This study does not involve Endangered or protected species according to IUZN (2021). The approval of sample collection is not required according to the Animal Ethical and Welfare Committee of Southwest Forestry University.
Author contributions
Hong Yu, Fen Wang, and Jin Xu conceived and designed the research; Hong Yu, Fen Wang and Jin Xu were involved in the analysis and interpretation of the data; Hong Yu and Fen Wang wrote the drafting of the paper and revised the manuscript; Jin Xu revised it critically for intellectual content; Jin Xu gave final approval of the version to be published. All the authors agreed to be listed and approved the submitted version of this manuscript.
Supplemental Material
Download TIFF Image (477.1 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI (https://www.ncbi.nlm.nih.gov) under accession no. OL711673. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA789424, SRR17245425, and SAMN24109441 respectively.
Additional information
Funding
References
- Aguila CP, Aikens RM, Ateliey PK, Buhr HM, Castro MG, Chua RJ, Dayal N, Deane HN, Dennehy B, Esenbekova M, et al. 2021. The complete mitochondrial genome of the Indian leafwing butterfly Kallima paralekta (insecta: lepidoptera: nymphalidae). Mitochondrial DNA B Resour. 6(1):274–277. doi: 10.1080/23802359.2020.1862000.
- Alexiuk MR, Lalonde MML, Marcus JM. 2021. Phylogenetic analysis of the complete mitochondrial genome of the Japanese peacock butterfly Aglais io geisha (Stichel 1907)(Insecta: lepidoptera: nymphalidae). Mitochondrial DNA B Resour. 6(10):3082–3084. doi: 10.1080/23802359.2021.1981168.
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477. doi: 10.1089/cmb.2012.0021.
- Boggs CL, Watt WB, Ehrlich PR. 2003. Butterflies: evolution and ecology taking flight. //Rocky Mountain Biological Lab Symposium Series. University of Chicago Press
- Brower AVZ. 2000. Phylogenetic relationships among the Nymphalidae (Lepidoptera) inferred from partial sequences of the wingless gene. Proc Biol Sci. 267(1449):1201–1211. doi: 10.1098/rspb.2000.1129.
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics. 10(1):1–9. doi: 10.1186/1471-2105-10-421.
- Chen K, Si C, Pan Z, Hao J. 2020. The complete mitochondrial genome of Aglais ladakensis (Lepidoptera: nymphalidae: nymphalinae). Mitochondrial DNA B Resour. 5(1):639–641. doi: 10.1080/23802359.2019.1711224.
- Chou IO. 1998. Classification and identification of Chinese butterflies. Zhengzhou: Henan Scientific and Technological Publishing House.
- Donath A, Jühling F, Al-Arab M, Bernhart SH, Reinhardt F, Stadler PF, Middendorf M, Bernt M. 2019. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 47(20):10543–10552. doi: 10.1093/nar/gkz833.
- Hamilton RV, Marcus JM, Lalonde MML. 2020. The complete mitochondrial genome of the black dead leaf butterfly Doleschallia melana (Insecta: lepidoptera: nymphalidae). Mitochondrial DNA Part B. 5(3):3306–3308. doi: 10.1080/23802359.2020.1814172.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780. doi: 10.1093/molbev/mst010.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649. doi: 10.1093/bioinformatics/bts199.
- Kemp DJ, Rutowski RL, Mendoza M. 2005. Colour pattern evolution in butterflies: a phylogenetic analysis of structural ultraviolet and melanic markings in North American sulphurs. Evol Ecol Res. 7(1):133–141.
- Kim MJ, Chu M, Park JS, Kim S-S, Kim I. 2021. Complete mitochondrial genome of the summer heath fritillary butterfly, Mellicta ambigua (Lepidoptera: nymphalidae). Mitochondrial DNA B Resour. 6(5):1603–1605. doi: 10.1080/23802359.2021.1917318.
- Lalonde MML. 2021. Phylogenetic analysis of the complete mitochondrial genome of the graphic beauty butterfly Baeotus beotus (Doubleday 1849)(Lepidoptera: nymphalidae: nymphalinae: coeini). Mitochondrial DNA Part B. 6(4):1516–1518. doi: 10.1080/23802359.2021.1914526.
- Li X-D, Ma Z-x, Rong W-T, Xu D, Li R. 2020. The complete mitochondrial genome of Polygonia c-aureum (Lepidoptera: nymphalidae) from Yizhou of China and its phylogeny. Mitochondrial DNA Part B. 5(3):2936–2937. doi: 10.1080/23802359.2020.1791759.
- Liu G, Chang Z, Chen L, He J, Dong Z, Yang J, Lu S, Zhao R, Wan W, Ma G, et al. 2020. Genome size variation in butterflies (Insecta, Lepidotera, Papilionoidea): a thorough phylogenetic comparison. Syst Entomol. 45(3):571–582. doi: 10.1111/syen.12417.
- Lohse K, Laetsch D, Vila R. 2021. The genome sequence of the large tortoiseshell, Nymphalis polychloros (Linnaeus, 1758). Wellcome Open Res. 6:238. doi: 10.12688/wellcomeopenres.17196.1.
- Price MN, Dehal PS, Arkin AP. 2010. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One. 5(3):e9490. doi: 10.1371/journal.pone.0009490.
- Schattner P, Brooks AN, Lowe TM. 2005. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33(Web Server issue):W686–W689. doi: 10.1093/nar/gki366.
- Shi Q-H, Sun X-Y, Wang Y-L, Hao J-S, Yang Q. 2015. Morphological characters are compatible with mitogenomic data in resolving the phylogeny of nymphalid butterflies (Lepidoptera: papilionoidea: nymphalidae). PLoS One. 10(4):e0124349. doi: 10.1371/journal.pone.0124349.
- Wu L-W, Lin L-H, Lees DC, Hsu Y-F. 2014. Mitogenomic sequences effectively recover relationships within brush-footed butterflies (Lepidoptera: nymphalidae). BMC Genomics. 15(1):468. doi: 10.1186/1471-2164-15-468.