
Abstract
Myrrophis (Enhydris) chinensis, also known as the Chinese water snake, has been used for medicinal purposes, such as the treatment of ailments involving fever, headache, and joint pain. The complete mitochondrial genome of M. chinensis was assembled using next-generation sequencing. The mitochondrial genome was 17,302 bp in length and contained 37 genes, including 13 protein-coding, 22 transfer RNA (tRNA), 2 ribosomal RNA genes, and 2 non-coding control regions (D-loop). The light chain of replication origin was found between tRNA-Asn and tRNA-Cys in the WANCY gene cluster, which is consistent with published mitogenomes of Homalopsidae. The phylogenetic tree supported the monophyly of Homalopsidae species and implied that M. chinensis is the closest related species to Myanophis thanlyinensis. The mitochondrial genome of M. chinensis provides fundamental data for exploring mitochondrial genome evolution in snakes (Homalopsidae).
Introduction
The Chinese water snake (Myrrophis chinensis) (Kumar et al. Citation2012) is an ovoviviparous colubrid snake widely distributed in Southern, Eastern, and Central China (including Anhui, Fujian, Guangdong, Guangxi, Hannan, Hunan, Hubei, Jiangsu, Jiangxi, Taiwan, and Zhejiang) (Zhao et al. Citation1998). M. chinensis is used in folk medicine to treat ailments such as fever, headache, and joint pain (Nóbrega et al. 2008). M. chinensis inhabits rice fields, ponds, and ditches that have important economic and scientific value. According to the ‘China Red Data Book of Endangered Animals’ and ‘China’s Red List of Biodiversity’, M. chinensis is considered a vulnerable species (Zhao et al. Citation1998; Wang et al. Citation2021). The assembly of M. chinensis mitogenome will facilitate research on species protection, delimitation, molecular evolution, and phylogenetic inference in snakes.
Materials and methods
All procedures were approved by the Animal Care and Use Committee of the Wenzhou University (Permit Number: WZU-049). A male adult M. chinensis was collected from Quzhou City, Zhejiang Province, China, in August 2022 (N 119°2′39.89″, E 28°52′36.77″) (). The muscle tissue was fixed with 95% ethanol and stored at −20 °C in the College of Life and Environmental Science, Wenzhou University (contact person: Yongpu Zhang, E-mail: [email protected]). The samples were transported to Genepioneer Biotechnologies Co. Ltd. (Nanjing, China) for genomic extraction and 150 bp paired-end library construction. Sequencing was performed using an Illumina NovaSeq 6000 sequencing platform (Illumina, USA). To verify the accuracy of the assembly, we aligned clean reads to the assembled genome to assess the depth of coverage. A total of 3220 reads were mapped to the complete genome sequence of My. thanlyinensis (NCBI GenBank ID: MW272554) in Bowtie 2 v2.2.4 (Langmead and Salzberg Citation2012), yielding 65 × coverage (supplemental Figure S1).
Figure 1. The specimen of Myrrophis chinensis from Quzhou City, Zhejiang Province, China (photo by Zhiwang Xu).

We used MITObim 1.9.1 (https://github.com/chrishah/MITObim) (Hahn et al. Citation2013) to assemble the M. chinensis mitochondrial genome using My. thanlyinensis as the reference. Additionally, a standard PCR method was used to amplify the mtDNA genes of M. chinensis for verification. Finally, we used tRNAscan-SE v.1.21 (http://lowelab.ucsc.edu/tRNAscan-SE) (Lowe and Chan Citation2016; Chan and Lowe Citation2019) and the MITOS web server (http://mitos2.bioinf.uni-leipzig.de/index.py) (Donath et al. Citation2019) to annotate the mitochondrial genome.
Results
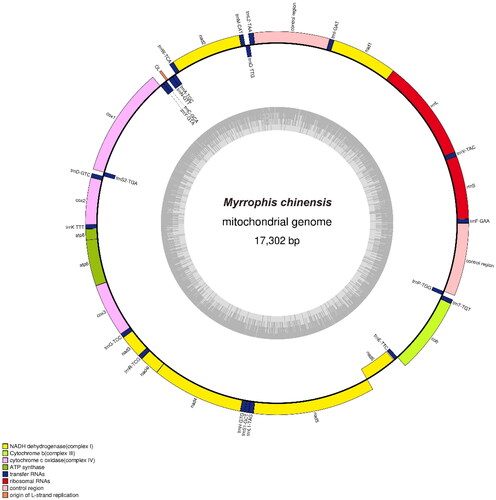
The mitochondrial genome of M. chinensis was 17,302 bp long (GenBank accession number: OP974098) () and consisted of 37 genes, including 22 transfer RNA (tRNA), 2 ribosomal RNA (16S and 12S), 13 protein-coding genes (PCGs), as well as 2 non-coding control regions (D-loop) and 1 light chain of replication origin (OL). Among the 13 PCGs, the longest and shortest were ND5 (1785 bp) and ATP8 (162 bp), respectively. The 12S and 16S rRNA genes were 922 and 1482 bp in length, respectively. One D-loop was located between tRNA-Ile and tRNA-Leu, with a length of 1111 bp, and the other was located between tRNA-Pro and tRNA-Phe, with a length of 1069 bp. The similarity between the two D-loops was 95.69%. The mitochondrial genome showed a relatively strong A and C bias, with base compositions of 33.23% A, 24.8% T, 28.7% C, and 13.26% G. Additionally, OL was located between tRNA-Asn and tRNA-Cys in the WANCY tRNA cluster region, which is consistent with the mitochondrial genomes of two known Homalopsidae species, My. thanlyinensis and Hypsiscopus (Enhydris) plumbea.
Figure 2. Gene map of the mitochondrial genome of Myrrophis chinensis. The forward orientation genes are located outside the circle, and the reverse orientation genes are located inside the circle. The inner gray circles represent GC content.
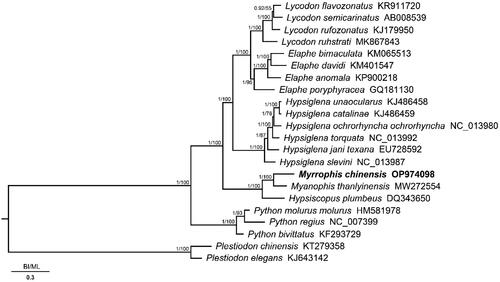
We constructed Bayesian inference (BI) implemented in MrBayes v.3.2.7a (Ronquist et al. Citation2012) and maximum likelihood (ML) implemented in RAxML v.8.2.12 (Stamatakis Citation2006) based on the 13 PCGs of M. chinensis and 21 related species to confirm the phylogenetic position of M. chinensis. The best-fitting substitution models and partitioning schemes were selected using PartitionFinder v.2.1.1 (Lanfear et al. Citation2017). Phylogenetic trees from the BI and ML analyses produced highly concordant topologies ().
Figure 3. Phylogenetic analysis of the 13 protein-coding genes of Myrrophis chinensis and 21 closely related species based on Bayesian inference (BI) and maximum likelihood (ML). Node numbers show Bayesian posterior probabilities from BI and bootstrap percentages from ML. GenBank accession numbers are given with species names, and the phylogenetic placement of M. chinensis is highlighted in bold.
Discussion and conclusion
In this study, we report the first complete mitogenome of M. chinensis established using high-throughput sequencing and assembly. The phylogenetic tree supported the monophyly of Homalopsidae, and M. chinensis showed the closest relationship to My. thanlyinensis (1.00 in BI and 100% in ML). The mitochondrial genome of M. chinensis provides fundamental data for exploring mitochondrial genome evolution in snakes (Homalopsidae).
Authors’ contributions
Yongpu Zhang contributed to the conception of the study; Jun Ping and Zhiwang Xu collected the samples and performed the research; Jun Ping analyzed the data and wrote the manuscript; and Yongpu Zhang revised the manuscript. All the authors have read and approved the final manuscript.
Ethical approval
All procedures were approved by the Animal Care and Use Committee of the Wenzhou University (Permit Number: WZU-049).
Supplemental Material
Download MS Word (113.1 KB)Supplemental Material
Download PNG Image (599.8 KB){kind=link}
Acknowledgments
We thank Lulu Shi for assistance in collecting samples during fieldwork. We are grateful to Dr. Jiayong Zhang (Zhejiang Normal University) for assistance with the mitochondrial genome assembly.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data supporting the findings of this study are available in the NCBI GenBank database (https://www.ncbi.nlm.nih.gov/) under accession no. OP974098. The associated BioProject, Sequence Read Archive (SRA), and Biosample numbers are PRJNA910348, SRR22586065, and SAMN32123642, respectively.
Additional information
Funding
References
- Chan PP, Lowe TM. 2019. tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol Biol. 1962:1–14. doi:10.1007/978-1-4939-9173-0_1.
- Donath A, Jühling F, Al-Arab M, Bernhart SH, Reinhardt F, Stadler PF, Middendorf M, Bernt M. 2019. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 47(20):10543–10552. doi:10.1093/nar/gkz833.
- Hahn C, Bachmann L, Chevreux B. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucleic Acids Res. 41(13):e129. doi:10.1093/nar/gkt371.
- Kumar AB, Sanders KL, George S, Murphy JC. 2012. The status of Eurostus dussumierii and Hypsirhina chinensis (Reptilia, Squamata, Serpentes): with comments on the origin of salt tolerance in homalopsid snakes. Syst Biodivers. 10(4):479–489. doi:10.1080/14772000.2012.751940.
- Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. 2017. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol. 34(3):772–773.
- Langmead B, Salzberg S. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9(4):357–359. doi:10.1038/nmeth.1923.
- Lowe TM, Chan PP. 2016. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44(W1):W54–W57. doi:10.1093/nar/gkw413.
- Nóbrega Alves RR, Silva Vieira WL, Santana GG. 2008. Reptiles used in traditional folk medicine: conservation implications. Biodivers Conserv. 17(8):2037–2049. doi:10.1007/s10531-007-9305-0.
- Ronquist F, Teslenko M, Mark PVD, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61(3):539–542. doi:10.1093/sysbio/sys029.
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 22(21):2688–2690. doi:10.1093/bioinformatics/btl446.
- Wang YZ, Cai B, Li JT. 2021. China’s red list of biodiversity: vertebrates (Volume III), Reptiles. Beijing: Science Press.
- Zhao EM, Huang MH, Zong Y. 1998. Fauna Sinica: Reptilia, Vol. Squamata: Serpentes. Beijing: Science Press.