
Abstract
Smallanthus sonchifolius (Asteraceae), is an important food plant in the world. There is no systematic report on the chloroplast genome of S. sonchifolius. Here we reported its complete chloroplast genome and analyzed the basic characteristics. The chloroplast genome was 152,301 bp in length, had a GC content of 37.55%, and encoded 113 unique genes, including 79 protein-coding genes, 4 ribosomal RNA genes, and 30 transfer RNA genes. Phylogenetic analysis showed that the tribe Millerieae and the tribe Madieae are closely related in the Asteraceae family. In the tribe Millerieae, Smallanthus was more closely related to Guizotia and Sigesbeckia. This chloroplast genome not only enriches the genome information of Smallanthus, but also lays the foundation for understanding the phylogeny within the genus Smallanthus.
Background
Smallanthus Mackenzie 1933 (Asteraceae, Millerieae), with about 24 species, occurs mainly from the southern United States to Central-Eastern Argentina. Some Smallanthus species can be important as medicinal species, among which Smallanthus sonchifolius (Poepp.) H. Rob. 1978 is a common medicinal and food plant in daily life (Aybar et al. Citation2001; Sánchez and Genta Citation2007). S. sonchifolius, also known as yacón, is a perennial herb that is native to the Andean region of South America. It is adaptable to different climates, altitudes and soils. The roots of S. sonchifolius are well hydrated and can usually be eaten raw (Ojansivu et al. Citation2011). Its roots also contain high levels of non-digestible oligosaccharides (NDOs), such as fructooligosaccharides (FOS) and inulin (Zardini Citation1991; Vos et al. Citation2007). High levels of FOS can reduce glycemic index, body weight and risk of colon cancer, which is important for human health (Choque Delgado et al. Citation2013).
There has been some progress in recent years regarding the phylogeny of Smallanthus. In 2009, researchers used phylogenetic analyses of nuclear (ITS) and plastid (matK) DNA sequences to preliminarily analyze the evolutionary relationships within Melampodium (Asteraceae, Millerieae) and also to preliminarily indicate the phylogenetic status of Smallanthus (Blöch et al. Citation2009). In 2014, a cluster analysis based on the morphology of all Smallanthus species was conducted to assess the monophyly and affinities among the Smallanthus species (Vitali and Viera Barreto Citation2014). In 2021, a phylotranscriptomic analysis based on more than 220 transcriptomic and few genomic data from 243 Asteraceae species yielded a highly supported phylogenetic tree of Asteraceae, which also indicated the phylogenetic position of S. sonchifolius. In 2022, the chromosome-level genome of S. sonchifolius was reported, clarifying the timing of divergence and recent polyploidy events (Fan et al. Citation2022).
Although many studies on the chloroplast genomes of Asteraceae species have been published in recent years, there is still a relative lack of chloroplast genome-related studies on S. sonchifolius, and its chloroplast genome characteristics are still poorly defined. Here, we assembled and analyzed the chloroplast genome of S. sonchifolius based on the short-read data. Our main objectives are as follows: (1) to analyze the structural features of the chloroplast genome of S. sonchifolius; (2) to analyze simple sequence repeats and long repeats; and (3) to validate the phylogenetic position of S. sonchifolius based on chloroplast genome.
Materials and methods
Plant material, DNA extraction and sequencing
Fresh leaves of S. sonchifolius were collected from Qujing, Yunnan, China (; 103.666036°E, 25.667044°N). The experimental material collected for this study species was conducted in accordance with institutional, national or international guidelines. The sample was deposited in the herbarium of the Forestry college, Xinyang Agriculture and Forestry University (voucher number: XLG001, Juan Yin, [email protected]). Whole genomic DNA was extracted using the CTAB method (Doyle and Doyle Citation1987). The DNA library of next generation sequencing with an insert size of 300 bp was constructed and sequenced using the Illumina Novoseq 6000 platform, yielding ∼6 Gb of raw data. The low-quality sequences were removed to obtain clean data using Trimmomatic v. 0.39 (Bolger et al. Citation2014).
Figure 1. Species reference images of S. sonchifolius (species images from Yin Juan, taken in a field in Qujing, Yunnan Province, 2022). (a) the leaves and flowers of S. sonchifolius; (b) and (c) the tubers of S. sonchifolius.

Genome assembly and annotation
De novo genome assembly from the clean data was accomplished using GetOrganelle v.1.7.5 (Jin et al. Citation2020). The parameters applied for plastome were ‘-R 15-k 21,45,65,85,105,127-F embplant_pt’. We used Bandage (Wick et al. Citation2015) to assess the integrity of the chloroplast genome assembly. Then we used samtools v1.7 (Li et al. Citation2009) and bedtools v2.28 (Quinlan and Hall Citation2010) for depth detection. The chloroplast genome was annotated by using CPGAVAS2 (Shi et al. Citation2019), PGA (Qu et al. Citation2019) and Geneious Prime v. 2022.2.2 with a reference genome (Sigesbeckia orientalis, GenBank: NC_053700). GB2sequin was then used to confirm the annotation results (Tillich et al. Citation2017). CPGView (http://www.1kmpg.cn/cpgview/) was used to visualize the chloroplast genome map and schematic diagrams of cis- and trans-splicing genes.
SSR and long repeat analysis
Simple sequence repeats (SSRs) were identified using the MISA software (https://webblast.ipk-gatersleben.de/misa/), including mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides with minimum numbers of 10, 5, 4, 3, 3, and 3, respectively (Beier et al. Citation2017). In addition, REPuter (https://bibiserv.cebitec.uni-bielefeld.de/reputer/) was used to calculate palindromic repeats, forward repeats, reverse repeats, and complementary repeats with the following settings: hamming distance of three and minimum repeat size of 30 bp (Kurtz et al. Citation2001).
Phylogenetic analysis
The chloroplast genomes of 39 Asteraceae species and one Campanulaceae species were downloaded from GenBank. A total of 41 species were involved in the phylogenetic analysis, and Campanula takesimana was used as an outgroup. We extracted 75 common protein-coding genes from the genome annotation files using PhyloSuite v. 1.2.2 (Zhang et al. Citation2020). Each protein-coding gene sequence was aligned using MAFFT v. 7.4 (Katoh and Standley Citation2013), and then the 75 aligned sequences were concatenated. Based on the matrix of concatenated sequences, a phylogenetic tree was constructed using the maximum likelihood (ML) method implemented in IQ-TREE v. 2.1.2 (Nguyen et al. Citation2015), and the best model was inferred using ModleFinder (Kalyaanamoorthy et al. Citation2017). Bootstrap analysis was performed with 1000 replicates. Tree visualization was achieved in Figtree v. 1.4.3 (https://github.com/rambaut/figtree/releases).
Results
General features of the chloroplast genome
We examined the chloroplast genome of S. sonchifolius for completeness, depth and GC content distribution, and the results showed that chloroplast genome was reliable (Figures S1 and S2). Chloroplast genome of S. sonchifolius was a typical circular tetrameric structure of 152,301 bp in length (; ; GenBank accession number: OQ295400), consisting of a large single copy (LSC) region (83,882 bp), a small single copy (SSC) region (18,351 bp), and a pair of inverted repeats (IR) (25,034 bp). The chloroplast genome encoded 113 unique genes, including 79 protein-coding genes, 4 ribosomal RNA genes and 30 transfer RNA genes (Table S1). The total GC content of the chloroplast genome was 37.55%, and the GC content of the IR region (43.08%) was significantly higher than that of the LSC region (35.61%) and the SSC region (31.30%).
Figure 2. The chloroplast genome map of S. sonchifolius. Genes on the inside of the circle are transcribed in a clockwise direction and genes on the outside of the circle are transcribed in a counter-clockwise direction. The light gray and dark gray shadows inside the inner circle indicated at and GC content, respectively.
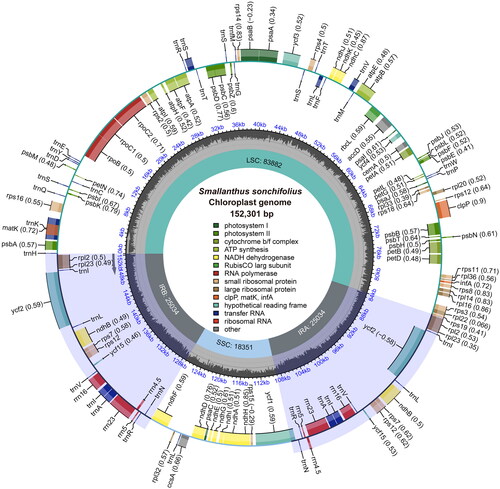
Table 1. Summary of the chloroplast genome of S. sonchifolius.
Repeat analysis
In the present study, we detected 144 SSRs in the chloroplast genome of S. sonchifolius (), including 33 mononucleotides, 39 dinucleotides, 21 trinucleotides, 26 tetranucleotides, 15 pentanucleotides and 10 hexanucleotides. Most of the SSRs were mononucleotides and dinucleotides, accounting for 50% of the total. In the chloroplast genome of S. sonchifolius, SSRs were most abundant in the LSC region and least abundant in the SSC region. In addition, we detected 50 long repeats (), including 16 forward repeats, 33 reverse repeats and 1 palindromic repeat. Most of them are forward and reverse repeats.
Phylogenetic analysis
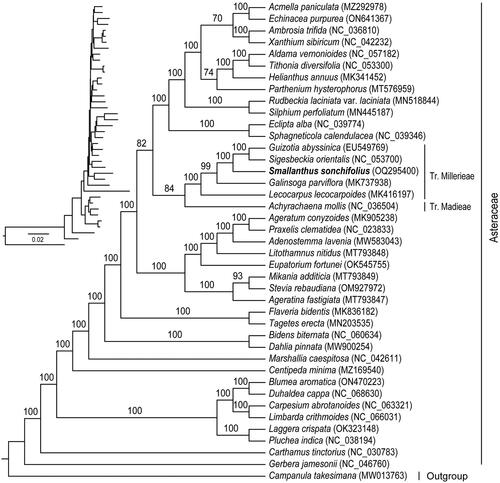
To understand the phylogenetic position of S. sonchifolius in the Asteraceae, we performed a phylogenetic analysis. The maximum-likelihood tree was constructed using IQ-TREE v. 2.1.2 (Nguyen et al. Citation2015) with the best-fit model of TVM + F+R4. The phylogenetic analysis showed 100% bootstrap support for all nodes in the phylogenetic tree, showing the reliability of the phylogeny (). Our phylogenetic tree clearly shows that the tribe Millerieae and the tribe Madieae were closely related, and that Smallanthus was more closely related to Guizotia and Sigesbeckia.
Figure 3. A maximum likelihood phylogenetic tree based on the concatenated sequences of 75 common protein-coding genes from 41 species. The number on the branch indicates the bootstrap value.
Discussion
Compared with nuclear genome and mitogenome, chloroplast genome was highly conserved and had been widely used in phylogenetic and evolutionary studies. Based on the chloroplast genome, the phylogenetic relationships of S. sonchifolius were analyzed. Despite the general inconsistency in the phylogeny of nuclear and organelle genes, our phylogenetic results were similar to those of previous phylogenetic studies based on low-copy nuclear genes (Zhang et al. Citation2021), both strongly supporting that Smallanthus was more closely related to Guizotia and Sigesbeckia, and indicating the high reliability of our results. However, the chloroplast genomes of other Smallanthus species are still unknown, and future genome sequencing of more Smallanthus species is needed to obtain more chloroplast genomes to study the phylogenetic relationships within the genus Smallanthus.
Conclusion
In this study, the chloroplast genome of S. sonchifolius was de novo assembled with short reads, and the chloroplast genome had a typical tetrameric structure similar to that of most angiosperms. The phylogenetic tree strongly supported the phylogenetic position of S. sonchifolius. The phylogenetic tree clearly showed that the tribe Millerieae and the tribe Madieae were closely related, and that Smallanthus was more closely related to Guizotia and Sigesbeckia. Thus, the chloroplast genome of S. sonchifolius not only enriches the genomic information of Smallanthus, but also lays the foundation for understanding the phylogeny within the genus Smallanthus.
Ethical approval
The S. sonchifolius is widely cultivated in China. Experimental studies do not include the genetic transformation, preservation of the genetic background of the species used, and any other processes requiring ethical approval. Therefore, no special permission is needed.
Authors’ contributions
Juan Yin was primarily responsible for the design of the experiment; Juan Yin, Zhen Wang and Guihua Ma participated in genome assembly and annotation work. Juan Yin, Wenjing Liu analyzed and interpreted the data. All authors have read and approved the final manuscript.
Supplemental Material
Download JPEG Image (464.8 KB){kind=link}
Supplemental Material
Download JPEG Image (200.7 KB){kind=link}
Supplemental Material
Download MS Excel (11.6 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The genome sequence data that support the findings of this study are available in GenBank of NCBI (http://www.ncbi.nlm.nih.gov/) under the accession no. OQ295400. The associated BioProject, BioSample, and SRA numbers are PRJNA932066, SAMN33145711, SRR23354904, respectively.
Additional information
Funding
References
- Aybar MJ, Sánchez Riera AN, Grau A, Sánchez SS., 2001. Hypoglycemic effect of the water extract of Smallantus sonchifolius (yacón) leaves in normal and diabetic rats. J Ethnopharmacol. 74(2):125–132. doi: 10.1016/s0378-8741(00)00351-2.
- Beier S, Thiel T, Münch T, Scholz U, Mascher M., 2017. MISA-web: a web server for microsatellite prediction. Bioinformat. 33(16):2583–2585. doi: 10.1093/bioinformatics/btx198.
- Blöch C, Weiss-Schneeweiss H, Schneeweiss GM, Barfuss MHJ, Rebernig CA, Villaseñor JL, Stuessy TF., 2009. Molecular phylogenetic analyses of nuclear and plastid DNA sequences support dysploid and polyploid chromosome number changes and reticulate evolution in the diversification of Melampodium (Millerieae, Asteraceae). Mol Phylogenet Evol. 53(1):220–233. doi: 10.1016/j.ympev.2009.02.021.
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30(15):2114–2120. doi: 10.1093/bioinformatics/btu170.
- Delgado GTC, Tamashiro WMdSC, Maróstica Junior MR, Pastore GM., 2013. Yacón (Smallanthus sonchifolius): a functional food. Plant Foods Hum Nutr. 68(3):222–228. doi: 10.1007/s11130-013-0362-0.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissues. Phytochem Bull. 19:11–15.
- Fan W, Wang S, Wang H, Wang A, Jiang F, Liu H, Zhao H, Xu D, Zhang Y., 2022. The genomes of chicory, endive, great burdock and yacón provide insights into Asteraceae palaeo-polyploidization history and plant inulin production. Mol Ecol Resour. 22(8):3124–3140. doi: 10.1111/1755-0998.13675.
- Jin J-J, Yu W-B, Yang J-B, Song Y, dePamphilis CW, Yi T-S, Li D-Z., 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):241. doi: 10.1186/s13059-020-02154-5.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS., 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589. doi: 10.1038/nmeth.4285.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780. doi: 10.1093/molbev/mst010.
- Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R., 2001. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29(22):4633–4642. doi: 10.1093/nar/29.22.4633.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R., 1000 Genome Project Data Processing Subgroup. 2009. The sequence alignment/map format and SAMtools. Bioinformat. 25(16):2078–2079. doi: 10.1093/bioinformatics/btp352.
- Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274. doi: 10.1093/molbev/msu300.
- Ojansivu I, Ferreira CL, Salminen S. 2011. Yacón, a new source of prebiotic oligosaccharides with a history of safe use. Trends Food Sci Technol. 22(1):40–46. doi: 10.1016/j.tifs.2010.11.005.
- Qu X-J, Moore MJ, Li D-Z, Yi T-S., 2019. PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods. 15(1):50. doi: 10.1186/s13007-019-0435-7.
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformat. 26(6):841–842. doi: 10.1093/bioinformatics/btq033.
- Sánchez S, Genta S. 2007. Yacón: un potencial producto natural para el tratamiento de la diabetes. Boletín Latinoamericano y del Caribe de. Plantas Medicinales y Aromáticas. 6(5):162–164.
- Shi L, Chen H, Jiang M, Wang L, Wu X, Huang L, Liu C., 2019. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47(W1):W65–W73. doi: 10.1093/nar/gkz345.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S., 2017. GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45(W1):W6–W11. doi: 10.1093/nar/gkx391.
- Vitali M, Viera Barreto J. 2014. Phylogenetic studies in Smallanthus (Millerieae, Asteraceae): a contribution from morphology. Phytotaxa. 159(2):77. doi: 10.11646/phytotaxa.159.2.2.
- Vos AP, van Esch BC, Stahl B, M'Rabet L, Folkerts G, Nijkamp FP, Garssen J., 2007. Dietary supplementation with specific oligosaccharide mixtures decreases parameters of allergic asthma in mice. Int Immunopharmacol. 7(12):1582–1587. doi: 10.1016/j.intimp.2007.07.024.
- Wick RR, Schultz MB, Zobel J, Holt KE., 2015. Bandage: interactive visualization of de novo genome assemblies. Bioinformat. 31(20):3350–3352. doi: 10.1093/bioinformatics/btv383.
- Zardini E. 1991. Ethnobotanical notes on “Yacón,” polymnia sonchifolia (Asteraceae). Econ Bot. 45(1):72–85. doi: 10.1007/BF02860051.
- Zhang C, Huang C-H, Liu M, Hu Y, Panero JL, Luebert F, Gao T, Ma H., 2021. Phylotranscriptomic insights into Asteraceae diversity, polyploidy, and morphological innovation. J Integr Plant Biol. 63(7):1273–1293. doi: 10.1111/jipb.13078.
- Zhang D, Gao F, Jakovlić I, Zou H, Zhang J, Li WX, Wang GT., 2020. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour. 20(1):348–355. doi: 10.1111/1755-0998.13096.