
Abstract
Deilephila elpenor is widely distributed in countries of Asia and Central Europe, and the larva is recognized as significant agriculture pest. The complete mitochondrial genome of D. elpenor is 15,372 bp in length. It contains 13 protein-coding genes (PCGs), 22 tRNA genes, two rRNA genes, and a control region. TAA is utilized as the termination codon for most PCGs, however, cox1 and cox2 use the incomplete termination codon T, and nad3 uses TAG as the termination codon. UUA (Leu) is the most frequently used codon, GCG (Ala) and CCG (Pro) are the least frequently used codons. In addition, we selected 15 species sequences closest to this species from NCBI, and used Manduca quinquemaculata and Manduca sexta (Lepidoptera: Smerinthinae) as the outgroup. Phylogenetic analysis suggested that D. elpenor was the most closely related to genus Theretra, genus Rhagastis and Cechenena minor.
Introduction
Deilephila elpenor (Linnaeus, 1758) is recognized as a kind of Lepidopteran pests that mainly affect flowers such as Impatiens balsamina, which are widely distributed in Asia and some countries in Central Europe, including China, Japan, South Korea, North Korea, and Russia. As a kind of nocturnal moth, the first evidence of animals with nocturnal color vision has been found on this species (Kelber et al. Citation2002). Due to the limited molecular data in GenBank database, there are little reports on the mitochondrial characteristics of this species and other species of the genus Deilephila. Therefore, we assembled and analyzed the complete mitochondrial genome of D. elpenor for the first time. The results would be able to provide molecular data for future studies of this species.
Materials
Adult moths of D. elpenor were collected in April 2021 in Mountain Dabieshan, Jinzhai county, Anhui Province, China (115°35′5″E, 31°41′32″N). The sample was alive during the collection. The specimen was deposited at the Entomological Museum, Anhui Normal University (Y-X Huang, [email protected]) under the voucher number AH2021042855. The identifying features of D. elpenor are as follows (): body color is pink, khaki and black; wingspan ranges from 52 to 75 mm; the upper forewing is khaki, and pink in the distal margin; the upper base of hindwing is black, and pink in the distal part; two longitudinal red bands on the sides of the head and thorax; the abdominal median line is red; both sides are khaki, and the outside is red. For details, please see https://tpittaway.tripod.com/china/d_elp.htm.
Figure 1. Morphological photograph of D. elpenor (photographed by Xiu-Shuang Zhu).

Methods
Total genomic DNA was extracted from legs of adult moths using the cetyltrimethylammonium bromide (CTAB) method (Shahjahan et al. Citation1995). 1 µg DNA was used for the DNA library preparations. Subsequently, the DNA libraries were sequenced using 150 bp paired reads on the Illumina NovaSeq 6000 platform (Illumina, San Diego, CA). FastQC was used to check the quality of data (Andrews Citation2020). A total of 43,338,253 clean reads were generated. Quality control standards are as follows: first, the reads with unrecognized nucleotides >10% were removed. Second, removing reads with >50% bases having Phred quality <5, and the readings aligned with the adapter greater than 10 nt were removed, allowing mismatches ≤10%. Finally, the assumed PCR repeats generated by PCR amplification during library construction were removed (the same readings 1 and 2 in the two paired end readings). The mitochondrial genome of D. elpenor was assembled using Novoplasty v2.7 (Nicolas et al. Citation2016). The depth of coverage was determined by Geneious (Kearse et al. Citation2012) by mapping the total clean reads to the newly assembled sequence (Figure S1). MITOS web server (Bernt et al. Citation2013) and MitoZ v3.0 (Meng et al. Citation2019) were utilized to annotate the complete mitogenome of D. elpenor. The drawing of mitogenome map was completed by CGView tool (Stothard and Wishart Citation2005). MEGA X was used to calculate the base composition and relative synonymous codon usage (RSCU) (Kumar et al. Citation2018).
To investigate the phylogenetic position of D. elpenor, we selected 15 species sequences closest to this species from NCBI, and used Manduca quinquemaculata and Manduca sexta (Lepidoptera: Smerinthinae) as the outgroup. Due to the relatively rapid evolution of the third codon site of protein-coding genes (PCGs), substitution saturation often occurs, which may hinder the reconstruction of phylogeny (Breinholt and Kawahara Citation2013). All sequences of the 13 PCGs were individually aligned in MEGA v10.1.8 and then concatenated into a PCG12 matrix (only the first and second codon positions) in DAMBE v5.3.74 (Huang et al. Citation2022). The maximum-likelihood (ML) tree was calculated with branch support estimated at 1000 bootstrap replications in IQ-Tree v1.4.1. Bayesian inference (BI) analysis was conducted using PhyloBayes (Lartillot et al. Citation2013). The BI tree is displayed in Figure S2.
Results
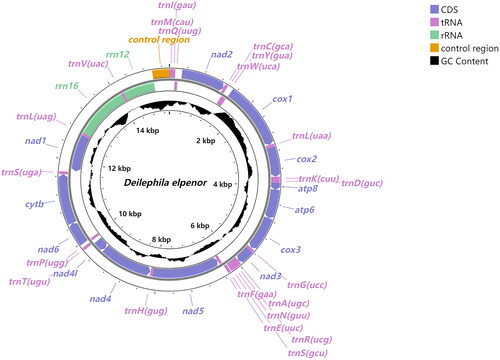
The full length of D. elpenor mitochondrial genome is 15,372 bp. It contains 37 genes (13 PCGs, 22 tRNA genes, and two rRNA genes) and a control region (). The mitogenome sequence has a high AT content of 81.2%. In addition, the majority coding strand (J-strand) contains 23 genes (nine PCGs and 14 tRNA genes) and the minority coding strand (N-strand) contains 14 genes (four PCGs, eight tRNA genes, and two rRNA genes). Analysis of the PCGs showed that the total length of the 13 PCGs was 11,198 bp, of which the nad5 gene was the longest of 1740 bp and atp8 was the shortest of 165 bp. TAA was utilized as the termination codon for most PCGs; however, cox1 and cox2 used the incomplete termination codon T, and nad3 used TAG as the termination codon. The analysis of rRNA genes showed that the length of 16S rRNA was 1368 bp with the AT content of 84.4%, and the length of 12S rRNA was 804 bp with the AT content of 84.8%. Besides, 184 bp of gaps and 24 bp of overlaps were detected in the mitogenome. The analysis of RSCUs showed that UUA (Leu) was the most frequently used codon, and GCG (Ala) and CCG (Pro) were the least frequently used codons.
Figure 2. Circular map of the mitochondrial genome of D. elpenor. GC content is represented by black, and other colors represent different gene types. Genes outside the circle are indicated to be encoded on the J-strand and those inside the circle are indicated to be encoded on the N-strand.
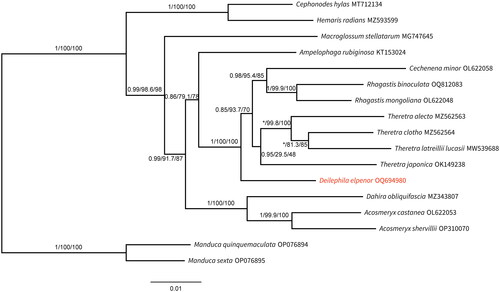
Both ML phylogenetic tree and BI analysis showed that the mitogenome of D. elpenor was the most closely related to Theretra alecto, Theretra clotho, Theretra latreillii lucasii, Theretra japonica, Rhagastis binoculata, Rhagastis mongoliana, and Cechenena minor with high branch support values ().
Figure 3. Based on the PCG12 matrix, the phylogenetic tree was reconstructed under the maximum-likelihood (ML) and Bayesian inference (BI) methods, with branch support values denoted PP (BI)/UFBoot2 (ML)/SH-Alrt (ML). The topology of ML tree is displayed and the nodes of BI tree that differ from ML tree are indicated by '*'. The mitochondrial genomes of 15 Macroglossinae species and two outgroup species of Smerinthinae (M. quinquemaculata and M. sexta) were selected. Mitochondrial genome sequences are derived from the following records: Cephonodes hylas MT712134, Hemaris radians MZ593599 (Huang et al. Citation2022); Macroglossum stellatarum MG747645 (Li et al. Citation2018); T. latreillii lucasii MW539688 (Lu et al. Citation2021); D. elpenor (this study); Ampelophaga rubiginosa KT153024, Cechenena minor OL622058, Rhagastis binoculata OQ812083, Rhagastis mongoliana OL622048, T. alecto MZ562563, T. clotho MZ562564, T. japonica OK149238, Dahira obliquifascia MZ343807, Acosmeryx castanea OL622053, Acosmeryx shervillii OP310070, M. quinquemaculata OP076894, and M. sexta OP076895 (unpublished).
Discussion and conclusions
In this study, the complete mitogenome of D. elpenor (GenBank accession no. OQ694980) was assembled and annotated for the first time. The complete mitochondrial genome was 15,372 bp in length and had an AT content of 81.2%, which was closest to Theretra latreillii lucasii (length: 15,354 bp) and Macroglossum stellatarum (AT content: 81.2%) in the Macroglossinae, respectively (Li et al. Citation2018; Lu et al. Citation2021). Codon usages showed that UUA (Leu) was the most frequently used codon, which was consistent with the results of Wang et al. (Citation2021) study on seven species of Macroglossinae. The mitochondrial tree reconstructed by both ML and BI method suggested that D. elpenor was the most closely related to genus Theretra, genus Rhagastis and Cechenena minor. Our studies demonstrate the structural features of the D. elpenor mitochondrial genome and explored its phylogenetic position in the Macroglossinae. The results add new species mitochondrial genomic data to the Macroglossinae and accumulate molecular data for the study of nocturnal moths. Besides, this is the first exploration of the phylogenetic position of the mitochondrial genome of the genus Deilephila species in the Macroglossinae.
Ethics statement
No specific permits were required for the insect specimens collected for this study. The field studies did not involve endangered or protected species. The insect species sequenced is a common hawkmoth species in China and is not included in the 'List of Protected Animals in China’.
Author contributions
Conception and design, analysis and interpretation of the data: XSZ, XW, and YFM. Drafting of the paper: XSZ and YXH. Revising it critically for intellectual content and the final approval of the version to be published: YXH. All authors agree to be accountable for all aspects of the work.
Supplemental Material
Download TIFF Image (2.6 MB)Supplemental Material
Download TIFF Image (448.3 KB)Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Data availability statement
The data that support the findings of this study are openly available in GenBank at https://www.ncbi.nlm.nih.gov/genbank/, reference number OQ694980. The associated BioProject, Bio-Sample numbers, and SRA are PRJNA948901, SAMN33923135, and SRR23974554, respectively.
Additional information
Funding
References
- Andrews S. 2020. FastQC: a quality control tool for high throughput sequence data; [accessed 2021 Sep 15]. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319. doi: 10.1016/j.ympev.2012.08.023.
- Breinholt JW, Kawahara AY. 2013. Phylotranscriptomics: saturated third codon positions radically influence the estimation of trees based on Next-Gen data. Genome Biol Evol. 5(11):2082–2092. doi: 10.1093/gbe/evt157.
- Huang Y-X, Xing Z-P, Zhang H, Xu Z-B, Tao L-L, Hu H-Y, Kitching IJ, Wang X. 2022. Characterization of the complete mitochondrial genome of eight diurnal hawkmoths (Lepidoptera: Sphingidae): new insights into the origin and evolution of diurnalism in sphingids. Insects. 13(10):887. doi: 10.3390/insects13100887.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649. doi: 10.1093/bioinformatics/bts199.
- Kelber A, Balkenius A, Warrant EJ. 2002. Scotopic colour vision in nocturnal hawkmoths. Nature. 419(6910):922–925. doi: 10.1038/nature01065.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549. doi: 10.1093/molbev/msy096.
- Lartillot N, Rodrigue N, Stubbs D, Richer J. 2013. PhyloBayes MPI: phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst Biol. 62(4):611–615. doi: 10.1093/sysbio/syt022.
- Li J, Zhang Y, Hu K, Zhao Y, Lin R, Li Y, Huang Z, Zhang X, Geng X, Ding J, et al. 2018. Mitochondrial genome characteristics of two Sphingidae insects (Psilogramma increta and Macroglossum stellatarum) and implications for their phylogeny. Int J Biol Macromol. 113:592–600. doi: 10.1016/j.ijbiomac.2018.02.159.
- Lu Q, Yao H, Zhang J, Xu H, Jiang C. 2021. The complete mitogenome sequence of the hawk moth, Theretra latreillii subsp. lucasii (Lepidoptera: Sphingidae) from Zhejiang Province, China. Mitochondrial DNA B Resour. 6(7):1880–1882. doi: 10.1080/23802359.2021.1934152.
- Meng G, Li Y, Yang C, Liu S. 2019. Mitoz: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 47(11):e63. doi: 10.1093/nar/gkz173.
- Nicolas D, Patrick M, Guillaume S. 2016. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 4(4):gkw955. doi: 10.1093/nar/gkw955.
- Shahjahan RM, Hughes KJ, Leopold RA, Devault JD. 1995. Lower incubation temperature increases yield of insect genomic DNA isolated by the CTAB method. Biotechniques. 19(3):332–334.
- Stothard P, Wishart DS. 2005. Circular genome visualization and exploration using CGView. Bioinformatics. 21(4):537–539. doi: 10.1093/bioinformatics/bti054.
- Wang X, Zhang H, Kitching I, Xu Z-B, Huang Y-X. 2021. First mitogenome of subfamily Langiinae (Lepidoptera: Sphingidae) with its phylogenetic implications. Gene. 789:145667. doi: 10.1016/j.gene.2021.145667.