
Abstract
Vanderbylia fraxinea (Bull.) D.A. Reid, 1973 is an important wood-inhabiting fungus that plays a significant role in nutrient recycling in most forest ecosystems. In this study, the complete mitochondrial genome of V. fraxinea was characterized through de novo assembly using Illumina sequencing data and genome annotation. The mitochondrial genome is a circular molecule of 115,473 bp with a GC content of 28.66%. It comprises a total of 62 genes. Among these, 36 are protein-coding genes including 21 free-standing open reading frames (ORFs), 24 transfer RNA genes, and two ribosomal RNA genes. Core gene set commonly found in fungal mitochondrial genomes is also present in this genome, such as the apocytochrome b (cob), three subunits of the cytochrome c oxidase (cox1, cox2, and cox3), seven subunits of the NADH dehydrogenase (nad1, nad2, nad3, nad4, nad4L, nad5, and nad6), and three subunits of the ATP synthase (atp6, atp8, and atp9), as well as ribosomal RNA subunits (rns and rnl) and a set of transfer RNA genes. Phylogenetic analysis of the protein-coding sequences from the mitochondrial genome revealed a close relationship between V. fraxinea and the Ganoderma species within the Polyporaceae family.
Introduction
The genus Vanderbylia D.A. Reid belongs to the phylum Basidiomycota, the class Agaricomycetes, the order Polyporales, and the family Polyporaceae (Kirk et al. Citation2001; Cui et al. Citation2019). It is a large, cosmopolitan genus typified by Polyporus medulla-panis (Jacq.) Fr., which is characterized by poroid basidiomata. It is a thick-walled, ellipsoid that distinctly truncates basidiospores and displays varying dextrinoid and amyloid reactions with a cyanophilous nature. The species within this genus exhibit a diverse range of ecological characteristics, functioning as saprophytes and either parasitic or pathogenic fungi (Dai et al. Citation2007, Citation2009; Younes et al. Citation2007; Wang et al. Citation2012). In most cases, this group possesses the ability to decompose cellulose, hemicellulose, and lignin within the plant cells as wood-inhabiting fungi, playing a significant role in nutrient recycling in most forest ecosystems (Dai Citation2012).
Vanderbylia fraxinea is either a parasite or a saprotroph growing on a broad range of hosts including Castanea, Fraxinus, Juglanas, Platanus, Populus, Prunus, Quercus, Robinia, Salix, and Ulmus (Kotlaba Citation1984; Kreisel Citation1987; Ryvarden and Gilbertson Citation1994). Among those within the genus Vanderbylia, it is considered an important group of fungi given their potential applications in bio-engineering. In particular, V. fraxinea can accumulate amounts of heavy metals, displaying high tolerance to Cd, Hg, and Cu. These characteristics position this fungus as a potential tool for monitoring environmental pollution (Sturini et al. Citation2017). Given the importance of this taxon, we investigated the phylogenetic relationship between V. fraxinea and the related genera based on the complete mitochondrial genome of the fungus.
Materials and methods
Samples of V. fraxinea were collected from Jinju, in the Gyeongsangnam province, South Korea (35°10′51.0″N 128°05′43.0″E) () and deposited to the culture collection (GNUFP) of Gyeongsang National University, South Korea (accession no. GNUFP287, gnu.ac.kr, Prof. Keumchul Shin, [email protected]). A voucher specimen was deposited to the herbarium of the Korea National Arboretum (KH; voucher no. KA23-0002C, kna.forest.go.kr, Dr. Chang Sun Kim, [email protected]). Genomic DNA was extracted from the isolate, GNUFP287, obtained from fungal mycelium harvested using fungal Maxwell 16 DNA purification kits (Promega, Madison, WI). An Illumina paired-end (PE) library was constructed and subsequently sequenced using the Illumina NovaSeq platform (San Diego, CA), producing 151 bp PE reads. Raw sequencing data of 3.4 Gb were generated and trimmed using the quality_trim program in the CLC Assembly Cell package ver. 4.2.1 (QIAGEN, Hvidovre, Denmark) with Phred scores >20. The trimmed data were used for the de novo assembly of the mitochondrial genome, as described in the previous studies (Lee et al. Citation2018; Cho et al. Citation2022, Citation2023). In brief, trimmed high-quality read sequences of 3.0 Gb were de novo assembled using the clc_novo_assemble program with default parameters in the CLC Assembly Cell. From the assembled contigs, mitochondrial contigs were selected and ordered through BLAST-based similarity searches (E-value cutoff 1e-5) against the mitochondrial sequences available in the NCBI organelle genome resources (https://www.ncbi.nlm.nih.gov/genome/organelle/). The selected mitochondrial contigs were merged and gap-filled through a series of read mapping and extension steps, resulting in the generation of a complete, circularized mitochondrial genome. Sequence error was investigated and subsequently corrected through read mapping of the trimmed sequence data and manual curation. The complete mitogenome sequence was annotated using the GeSeq (Tillich et al. Citation2017) and Artemis (Carver et al. Citation2012) programs with reference mitochondrial genomes (GenBank accession nos. KR109212.1, KP410262.1, MN356101.1, MT843218.1, and NC_021750.1). In addition, gene annotation was reconfirmed through manual curation, involving BLAST searches against the reference mitochondrial genomes.
Conserved protein-coding sequences were identified among the mitochondrial genomes of V. fraxinea and related 17 species and concatenated for each species. The concatenated sequences were multiple-aligned using MAFFT ver. 7 (http://mafft.cbrc.jp/alignment/server/index.html) (Katoh et al. Citation2019) with default parameters and used as input data for phylogenetic analysis. Phylogenetic analysis was performed using a Bayesian inference method of BEAST2 ver. 2.6.7 (Bouckaert et al. Citation2019), with default parameters (Site Substitution Model = Gamma Site Model (Gamma Category = 4; GTR), Chain length of MCMC = 10,000,000, Burn-in = 10%, Model = Yule model).
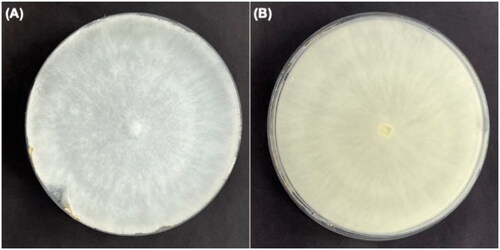
Figure 1. Colony morphology of the isolate of Vanderbylia fraxinea, GNUFP287, on the potato dextrose agar (PDA); (A) obverse and (B) reverse. The photo provided by Prof. Shin, Keumchul, was taken during cultivation of V. fraxinea after collection made from Prunus × yedoensis.
Results and discussion
The mitochondrial genome of V. fraxinea is a circular molecule of 115,473 bp consisting of 28.66% GC content (GenBank accession no. OQ612670) and a ×2948 mean coverage of trimmed sequencing data (). In total, 62 genes were predicted in this mitochondrial genome comprising 36 protein-coding genes, which included 21 free-standing open reading frames (ORFs), 24 transfer RNA genes, and two ribosomal RNA genes (). Core genes commonly found in fungal mitochondrial genomes are also identified, which include the apocytochrome b (cob), three subunits of the cytochrome c oxidase (cox1, cox2, and cox3), seven subunits of the NADH dehydrogenase (nad1, nad2, nad3, nad4, nad4L, nad5, and nad6), and three subunits of the ATP synthase (atp6, atp8, and atp9), as well as untranslated genes of the small and large ribosomal RNA subunits (rns and rnl, respectively) and 24 transfer RNA (trnA-UGC to trnY-GUA) genes. Most genes consist of a single exon, but four genes consist of multiple exons, such as cox1 (16 exons and 15 introns), nad4 (three exons and two introns), rnl (three exons and two introns), and rns (two exons and one intron).
Figure 2. Mitochondrial genome map of Vanderbylia fraxinea. The map was prepared using OGDRAW program (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html) (Lohse et al. Citation2007). Genes transcribed clockwise and counterclockwise are indicated on the outside and inside of the large circle, respectively. GC contents are indicated on the inner circle. This genome map does not show ORFs.
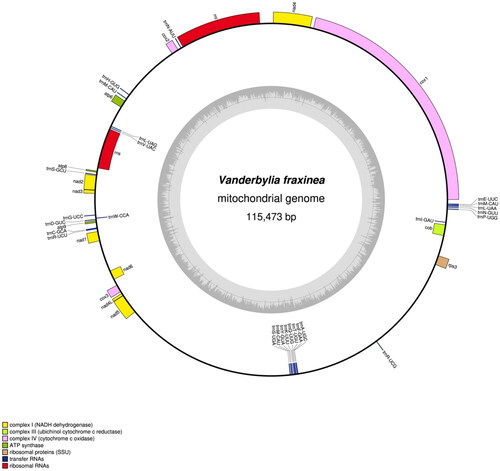
Table 1. The annotated gene list of mitochondrial genome in Vanderbylia fraxinea.
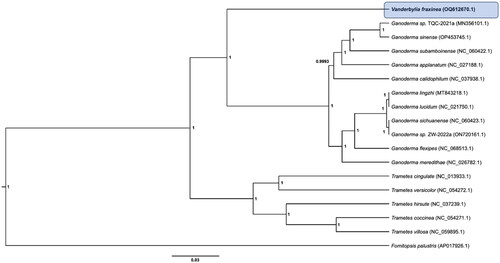
A phylogenetic tree based on a Bayesian inference method with 13 conserved protein-coding sequences of atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad5, and nad6 revealed that V. fraxinea is close to the Ganoderma species in the Polyporaceae family ().
Figure 3. Phylogenetic tree of mitochondrial genomes of Vanderbylia fraxinea and its related species. Protein-coding sequences conserved in the mitochondrial genomes of 18 species were multiple-aligned using MAFFT (http://mafft.cbrc.jp/alignment/server/index.html) and used to generate phylogenetic tree using Bayesian inference method of BEAST2 (Bouckaert et al. Citation2019) with default parameters. Posterior probability values are on the branches. The isolate, V. fraxinea (GNUFP287), obtained in this study was represented by bold letters. GenBank accession nos. of mitochondrial genome sequences used for this tree are indicated within parentheses.
The complete mitochondrial genome sequence generated in this study will provide valuable genetic information for understanding the phylogenetic relationship of this species at the molecular level.
Ethics statement
Ethical approval for the study was obtained from the National Institute of Forest Science, South Korea. The collection and analysis of plant material were performed in accordance with guidelines provided by the National Institute of Forest Science, South Korea.
Authors contributions
Sung-Eun Cho and Dong-Hyeon Lee conceived the research project and drafted the manuscript. Sanggon Lee, Misong Kim, and Keumchul Shin contributed to cultivation management and collection of fungal samples. Sung-Eun Cho, Dong-Hyeon Lee, Heonil Kang, Sang-Tae Seo, Namkyu Kim, and Keumchul Shin performed the data analysis and revised the manuscript critically for intellectual content. All authors contributed to the final approval of the version to be published and all agree to be accountable for all aspects of the work.
Acknowledgements
The authors thank Kyunghyun Nam in Phyzen Co. for assistance in bioinformatics analyses.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov under the accession no. OQ612670. The associated BioProject, Bio-Sample, and SRA numbers are PRJNA943535, SAMN33730946, and SRR23823732, respectively.
Additional information
Funding
References
- Bouckaert R, Vaughan TG, Barido-Sottani J, Duchêne S, Fourment M, Gavryushkina A, Heled J, Jones G, Kühnert D, De Maio N, et al. 2019. BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 15(4):e1006650. doi:10.1371/journal.pcbi.1006650.
- Carver T, Harris SR, Berriman M, Parkhill J, McQuillan JA. 2012. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics. 28(4):464–469. doi:10.1093/bioinformatics/btr703.
- Cho SE, Oh JY, Lee DH. 2022. Complete mitochondrial genome sequence of Colletotrichum siamense isolated in South Korea. Microbiol Resour Announc. 11(5):e01055-21. doi:10.1128/mra.01055-21.
- Cho SE, Oh JY, Lee DH. 2023. The complete mitochondrial genome of Cladosporium anthropophilum (Cladosporiaceae, Dothideomycetes). Mitochondrial DNA B Resour. 8(1):164–166. doi:10.1080/23802359.2023.2167474.
- Cui BK, Li HJ, Ji X, Zhou JL, Song J, Si J, Yang ZL, Dai YC. 2019. Species diversity, taxonomy and phylogeny of Polyporaceae (Basidiomycota) in China. Fungal Diversity. 97(1):137–392. doi:10.1007/s13225-019-00427-4.
- Dai YC, Cui BK, Yuan HS, Li BD. 2007. Pathogenic wood‐decaying fungi in China. For Pathol. 37(2):105–120. doi:10.1111/j.1439-0329.2007.00485.x.
- Dai YC, Yang ZL, Cui BK, Yu CJ, Zhou LW. 2009. Species diversity and utilization of medicinal mushrooms and fungi in China. Int J Med Mushr. 11(3):287–302. doi:10.1615/IntJMedMushr.v11.i3.80.
- Dai YC. 2012. Polypore diversity in China with an annotated checklist of Chinese polypores. Mycoscience. 53(1):49–80. doi:10.1007/s10267-011-0134-3.
- Katoh K, Rozewicki J, Yamada KD. 2019. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 20(4):1160–1166. doi:10.1093/bib/bbx108.
- Kirk PM, Cannon PF, David JC, Stalpers JA. 2001. Ainsworth and Bisby’s dictionary of the fungi. 9th ed. Wallingford (UK): CAB International.
- Kotlaba F. 1984. Geographical distribution and ecology of polypores (Polyporales s.l.) in Czechoslovakia (in Czech). Academia, Praha, p. 1–194.
- Kreisel H. 1987. Pilzfl ora der Deutschen Demokratischen Republik. Basidiomycetes (Gallert-, Hut- und Bauchpilze). Jena: Germany: G. Fischer Verlag.
- Lee HO, Choi JW, Baek JH, Oh JH, Lee SC, Kim CK. 2018. Assembly of the mitochondrial genome in the Campanulaceae family using Illumina low-coverage sequencing. Genes. 9(8):383. doi:10.3390/genes9080383.
- Lohse M, Drechsel O, Bock R. 2007. OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet. 52(5–6):267–274. doi:10.1007/s00294-007-0161-y.
- Ryvarden L, Gilbertson RL. 1994. European polypores. Part 2. Meripilus-Tyromyces. Oslo, Norway: Fungiflora; p. 393–743.
- Sturini M, Girometta C, Maraschi F, Savino E, Profumo A. 2017. A preliminary investigation on metal bioaccumulation by Perenniporia fraxinea. Bull Environ Contam Toxicol. 98(4):508–512. doi:10.1007/s00128-017-2038-1.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq-Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45(W1):W6–W11. doi:10.1093/nar/gkx391.
- Wang W, Yuan T, Wang K, Cui B, Dai Y. 2012. Combination of biological pretreatment with liquid hot water pretreatment to enhance enzymatic hydrolysis of Populus tomentosa. Bioresour Technol. 107:282–286. doi:10.1016/j.biortech.2011.12.116.
- Younes SB, Mechichi T, Sayadi S. 2007. Purification and characterization of the laccase secreted by the white rot fungus Perenniporia tephropora and its role in the decolourization of synthetic dyes. J Appl Microbiol. 102:1033–1042.