
Abstract
The family Orchidaceae is renowned for its extensive diversity. Within this family, the genus Goodyera R. Br. is classified under the subtribe Goodyerinae, comprising approximately 99 species. In this study, a species Goodyera yunnanensis Schltr., its plastid genome was characterized. The plastid genome of G. yunnanensis is 146,197 bp in size and exhibits a typical quadripartite structure with a pair of inverted repeat regions (IRs) of 25,611 bp, a large single-copy region (LSC) of 81,300 bp and a small single-copy region (SSC) of 13,675 bp. A total of 126 genes were identified, containing 80 protein-coding genes, 38 tRNA genes and 8 rRNA genes. The overall GC content is 37.2%, with corresponding values of 43.3%, 34.7% and 29.1% in IR, LSC and SSC regions, respectively. Forty-seven simple sequence repeats (SSRs) are found in G. yunnanensis plastome, and the frequency of mononucleotide repeats is significantly higher than other repeat types. Phylogenetic analysis indicates that Goodyera is resolved into four clades. G. yunnanensis belongs to the monophyletic clade A, and its phylogenetic position can be reasonably supported by morphological and molecular data.
Introduction
The family Orchidaceae is renowned for its extensive diversity, encompassing around 736 recognized genera and 28,000 species worldwide (Christenhusz and Byng Citation2016). Within this family, the genus Goodyera R. Br. is classified under the subtribe Goodyerinae, comprising approximately 99 species, and is in Asia, North America, Europe, Southern Africa, Northeast Australia, Madagascar, and the Pacific Islands (Kim and Kim Citation2022). The vast majority of Goodyera species are terrestrial, growing on moist grounds or mossy rocks in mountains, characterized by leaves with white or golden reticulate veins, elongate and creeping rhizomes, hoods including dorsal sepals and petals, resupinate flowers, and concave-saccate labellum.
The plants of Goodyera have significant ornamental and medicinal values, but some unresolved issues of phylogeny are still retained within the genus. Shin et al. (Citation2002) used the nuclear ribosomal internal transcribed spacer (nrITS) region to reveal the monophyly of Korean Goodyera species. Juswara (Citation2010) elucidated the phylogenetic relationships of multiple Goodyera species based on the nrITS and chloroplast fragments (trnL-F and rpl16), whereas Goodyera was demonstrated to be polyphyletic in the study. Later, Smidt et al. (Citation2020) inferred that Goodyera was biphyletic by carrying out the comparative analysis of plastid genomes. Although different studies have been conducted, the monophyly of Goodyera in East Asia and North America has yet to be clarified.
Goodyera yunnanensis Schltr. (1919) is a member of this genus, which has typically narrow-ovate sepals, oblong-ligulate and oblique petals, and a minutely papillose lip epichile. To date, no molecular data are available for resolving the phylogenetic position of G. yunnanensis, and mining the genetic information of this species is undoubtedly extremely important for refining the phylogenetic framework of Goodyera. Therefore, we characterize the plastid genome of G. yunnanensis here, which will provide valuable plastome information for the further classification, evolution, and conservation of genetic diversity in Goodyera.
Materials and methods
G. yunnanensis () in this study was sampled from Yunnan Province, China (28°30′56.20″N, 99°57′41.62″E). Voucher specimen (DT096-10) was deposited in the herbarium of the Kunming Institute of Botany (KIB, Kunming, China) (Tao Deng, [email protected]). Total genomic DNA was extracted from the silica-dried leaf by using a modified CTAB method, and its DNA material was also deposited in the herbarium of KIB. The whole genome was sequenced by Novogene Technologies Co. Ltd. (Beijing, China) on the Illumina Hiseq platform. In total, 1.15 G of the clean data was used to assemble the plastid genome sequence on the GetOrganelle program (Jin et al. Citation2018) with the following parameters: R-10; t-1; k-75, 95, 115, 127. The coverage depth was calculated by mapping readings onto plastid genome sequences using bowtie2 to determine the correctness of assembly (Langmead and Salzberg Citation2012). Then, the genome sequence was annotated on website Geseq (https://chlorobox.mpimp-golm.mpg.de/geseq.html) (Tillich et al. Citation2017), and annotation results were manually checked and corrected in software Geneious-9.0.2 with Goodyera biflora (Lindl.) Hook.f. (Genbank: OM314910) as a reference. Finally, the annotated plastid genome of G. yunnanensis was deposited in the GenBank database under the accession no. OQ935753. The Organellar Genome DRAW (OGDRAW) and Chloroplast Genome Viewer (CPGView) were used to draw circular map of the plastid genome and to annotate genes, respectively. To calculate simple sequence repeats (SSRs) of G. yunnanensis plastid genome using MISA online tool, the parameters were set to 10, 5 and 4 repeats for mononucleotide, dinucleotide and trinucleotide, and 3 repeats were used for tetranucleotide, pentanucleotide and hexanucleotide, respectively (Li and Wan Citation2005). For phylogenetic analysis, referring to Kim and Kim (Citation2022) study, plastid genome sequences of 26 species were aligned based on MAFFT method (Katoh and Standley Citation2013), and Ophrys sphegodes Mill. (Genbank: AP018717) was chosen as an outgroup for constructing ML phylogenetic tree. The ML tree was constructed by using RAxML-8.2.10 under the GTRGAMMA model with 1,000 bootstrap replicates on Cipres Portal (https://www.phylo.org/portal2) (Stamatakis Citation2014).
Figure 1. The living plant of Goodyera yunnanensis used in this study. This species has typically narrow-ovate sepals, oblong-ligulate and oblique petals. The photograph was taken by Mei-Xiang Hu in Shangri-La, Yunnan, China, and was used with the author’s permission.

Results
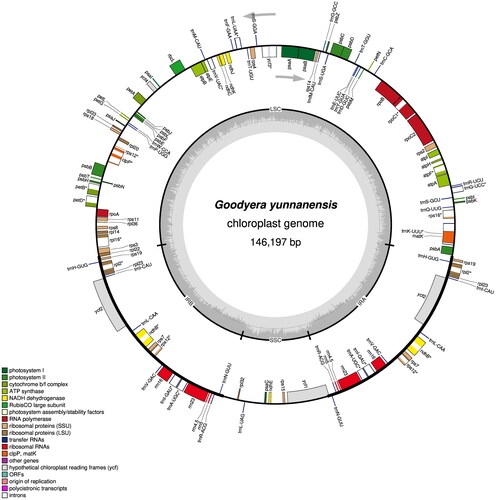
The plastid genome of G. yunnanensis () was 146,197 bp in size with an average coverage of 1,264× (Figure S1) and exhibited a typical quadripartite structure with a pair of inverted repeat regions (IRs) of 25,611 bp, a large single-copy region (LSC) of 81,300 bp and a small single-copy region (SSC) of 13,675 bp. A total of 126 genes were identified, containing 80 protein-coding genes, 38 tRNA genes and 8 rRNA genes. Ten cis-splicing genes including rps16, atpF, rpoC1, ycf3, clpP, petB, petD, rpl16, rpl2 and ndhB, and one trans-splicing genes rps12 were detected (Figure S2). The overall GC content was 37.2%, with corresponding values of 43.3%, 34.7% and 29.1% in IR, LSC and SSC regions, respectively. Forty-seven simple sequence repeats (SSRs) were found in the plastome with 28 mononucleotide (A-5, T-23), 8 dinucleotide (AT-4, TA-2, TC-1, GA-1), 3 trinucleotide (ATA-1, CTA-1, TCT-1), 6 tetranucleotide (AGAA-1, CATT-1, GATA-1, GTCT-1, TCTT-1, TTGA-1), and 2 pentanucleotide repeats (TTATT-1, TTCTT-1). The frequency of mononucleotide repeats was significantly higher than other repeat types, accounting for approximately 59.5% of the total SSRs (Table S1).
Figure 2. Gene map of the plastid genome of Goodyera yunnanensis was drawn using Organellar genome DRAW (OGDRAW). the center of the map indicates the species name and size of the plastid genome. The outer circle represents the gene, showing the direction of transcription of the gene, while the inner circle represents the size of the IRs, LSC, and SSC, respectively. Genes belonging to different functional groups have been color-coded. The darker gray area in the inner circle corresponds to GC content, and the gray line in Middle represents the 50% threshold, whereas the lighter gray corresponds to at content.
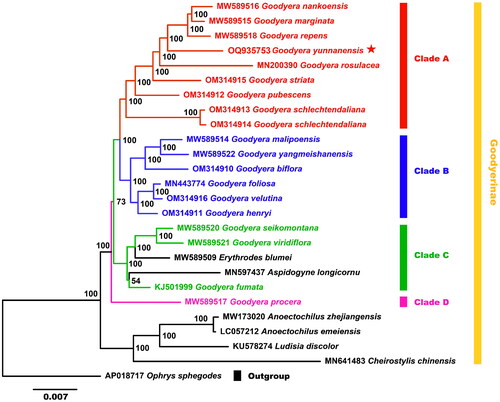
The matrix of 26 plastid genome sequences contained 169,224 bp, and the ML tree presented four major clades (A, B, C and D). The ML bootstrap value of most nodes was 100%, with only two nodes having weaker bootstrap value (). Our phylogenetic analysis result revealed that Goodyera was resolved into four clades. G. yunnanensis formed a monophyletic clade A with G. nankoensis Fukuy., G. marginata Lindl., G. repens (L.) R.Br., G. rosulacea Y.N.Lee, G. striata Rchb.f., G. pubescens (Willd.) R.Br. and G. schlechtendaliana Rchb.f. Clade B was also monophyletic with six Goodyera species, sister to the clade A. Clade C comprises three species from Goodyera, which were nested with Erythrodes blumei (Lindl.) Schltr. and Aspidogyne longicornu (Cogn.) Garay. Clade D was represented only by G. procera (Ker Gawl.) Hook.
Figure 3. Phylogenetic relationships among 18 Goodyera species and other species of Goodyerinae were inferred from ML analyses under the GTRGAMMA based on the whole plastid genome, with Ophrys sphegodes as the outgroup, and the phylogenetic position of Goodyera yunnanensis (OQ935753) is marked with a red asterisk. The bootstrap support values are exhibited next to the nodes, and the Genbank accession numbers of each genome are shown in front of the latin name, respectively. The plastid genome sequences used for phylogenetic reconstruction were all referenced and derived from Kim and Kim (2002) study, as follows: Goodyera nankoensis MW589516, G. marginata MW589515, G. repens MW589518, G. rosulacea MN200390, G. striata OM314915, G. pubescens OM314912, G. schlechtendaliana OM314913/OM314914, G. malipoensis MW589514, G. yangmeishanensis MW589522, G. biflora OM314910, G. foliosa MN443774, G. velutina OM314916, G. henryi OM314911, G. seikomontana MW589520, G. viridiflora MW589521, G. fumata KJ501999, G. procera MW589517, Erythrodes blumei MW589509, Aspidogyne longicornu MN597437, anoectochilus zhejiangensis MW173020, A. emeiensis LC057212, ludisia discolor KU578274, cheirostylis chinensis MN641483, Ophrys sphegodes AP018717.
Disscussion
We assembled the plastid genome of G. yunnanensis, which showed a quadripartite structure with 126 genes, 80 protein-coding genes and 46 RNA genes. However, we observed that this species lost some genes (ndhD, ndhG, ndhI, ndhA) related to NADPH-quinone oxidoreduction, and the deletion of these types of genes was also common in angiosperms (Mohanta et al. Citation2020). There were Forty-seven SSRs detected in G. yunnanensis plastid genome. The A or T mononucleotide repetition is the most primary repetitive type, and all mononucleotide repeats are composed of A and T. This distribution pattern of SSR is consistent with previous study that A and T mononucleotide are the most abundant repeats in the plastid genome of most angiosperms, and rarely include tandem G or C repeats (Kuang et al. Citation2011). The SSR exhibited polymorphic and codominance genetic patterns. These sequences have been widely used to speculate genetic variation between plant genotypes and served as DNA markers in population genetic studies (Deguilloux et al. Citation2004). Our phylogenetic result revealed that Goodyera was resolved into four clades with similar topology to Kim and Kim (Citation2022), and G. yunnanensis was integrated into the clade A with strong support. The formation of clade A can also be supported by morphology. That is, except for G. schlechtendaliana, the remaining species have the common feature of being glabrous inside the labellum. Additionally, G. procera was identified as the independent the clade D, with distinctive characters such as narrowly ovate-elliptic leaves or densely non-secund flowers. The clade B includes six species of Goodyera, which share the silver or gold veins and closed lateral sepals (Hu et al. Citation2016; Kim and Kim Citation2022). The clade C is a non-monophyletic clade, containing three Goodyera species, as well as E. blumei and A. longicornu, while the nesting of the latter two was also found in previous studies based on DNA fragments (nrITS, trnL-F, and matK) and plastid-coding genes analyses (Hu et al. Citation2016; Tu et al. Citation2021; Kim and Kim Citation2022). Therefore, to thoroughly clarify the phylogenetic relationships within Goodyera, it is necessary to sample more taxa and conduct more in-depth researches.
Conclusion
We reported plastid genome of G. yunnanensis, revealing its genome structure, sequence composition, and the phylogenetic relationship between this species and its related taxa. The relationship also received reasonable support from morphological and molecular data. Our study results will provide valuable information for further study on taxonomy, identification and evolution of Goodyera.
Author contributions
Meixiang Hu and Guiyun Huang planned and designed the research. Xiongying Wang and Jingyi Peng collected the plant materials. Haofei Zhu, Xiongying Wang, Huiyuan Chen, Pianpian Li, Jun Zhang and Jingyi Peng performed experiments. Jingyi Peng analyzed the data. Haofei Zhu wrote the manuscript. All authors agree to be accountable for all aspects of the work.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors. In this experiment, we did not collect any human or animal samples.
Supplemental Material
Download MS Excel (11.6 KB)Supplemental Material
Download MS Word (1.9 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov/ under the GenBank accession OQ935753. The associated BioProject, SRA and BioSample numbers are PRJNA970196, SRR24464625 and SAMN34995382, respectively.
Additional information
Funding
References
- Christenhusz MJ, Byng JW. 2016. The number of known plants species in the word and its annual increase. Phytotaxa. 261(3):201–217. doi:10.11646/phytotaxa.261.3.1.
- Deguilloux MF, Pemonge MH, Petit RJ. 2004. Use of chloroplast microsatellites to differentiate oak populations. Ann for Sci. 61(8):825–830. doi:10.1051/forest:2004078.
- Hu C, Tian HZ, Li HQ, Hu AQ, Xing FW, Bhattacharjee A, Hsu TC, Kumar P, Chung SW. 2016. Phylogenetic analysis of a‘jewel orchid’genus Goodyera (Orchidaceae) based on DNA sequence data from nuclear and plastid regions. PLoS One. 11(2):e0150366. doi:10.1371/journal.pone.0150366.
- Jin JJ, Yu WB, Yang JB, Song Y, Yi TS, Li DZ. 2018. GetOrganelle: a simple and fast pipeline for de novo assembly of a complete circular chloroplast genome using genome skimming data. BioRxiv. 867.
- Juswara LS. 2010. Phylogenetic analyses of subtribe Goodyerinae and revision of Goodyera section Goodyera (Orchidaceae) from Indonesia, and fungal association of Goodyera section Goodyera. [Doctoral dissertation. OhioLINK Electronic Theses and Dissertations Center]. Columbus (OH): Ohio State University. http://rave.ohiolink.edu/etdc/view?acc_num=osu1275490522.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780. doi:10.1093/molbev/mst010.
- Kim TH, Kim JH. 2022. Molecular phylogeny and historical biogeography of Goodyera R. Br. (Orchidaceae): a case of the vicariance between East Asia and North America. Front Plant Sci. 13:850170. doi:10.3389/fpls.2022.850170.
- Kuang DY, Wu H, Wang YL, Gao LM, Zhang SZ, Lu L. 2011. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): implication for DNA barcoding and population genetics. Genome. 54(8):663–673. doi:10.1139/g11-026.
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9(4):357–359. doi:10.1038/nmeth.1923.
- Li Q, Wan JM. 2005. SSRHunter: development of a local searching software for SSR sites. Yi Chuan. 27(5):808–810.
- Liu S, Ni Y, Li J, Zhang X, Yang H, Chen H, Liu C. 2023. CPGView: a package for visualizing detailed chloroplast genome structures. Mol Ecol Resour. 23(3):694–704. doi:10.1111/1755-0998.13729.
- Mohanta TK, Mishra AK, Khan A, Hashem A, Abd Allah EF, Al-Harrasi A. 2020. Gene loss and evolution of the plastome. Genes (Basel). 11(10):1133. doi:10.3390/genes11101133.
- Shin KS, Shin YK, Kim JH, Tae KH. 2002. Phylogeny of the genus Goodyera (Orchidaceae; Cranichideae) in Korea based on nuclear ribosomal DNA ITS region sequences. J Plant Biol. 45(3):182–187. doi:10.1007/BF03030312.
- Smidt EC, Páez MZ, Vieira LN, Viruel J, de Baura VA, Balsanelli E, de Souza EM, Chase MW. 2020. Characterization of sequence variability hotspots in Cranichideae plastomes (Orchidaceae, Orchidoideae). PLoS One. 15(1):e0227991. doi:10.1371/journal.pone.0227991.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313. doi:10.1093/bioinformatics/btu033.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq-versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45(W1):W6–W11. doi:10.1093/nar/gkx391.
- Tu XD, Liu DK, Xu SW, Zhou CY, Gao XY, Zeng MY, Zhang S, Chen JL, Ma L, Zhou Z, et al. 2021. Plastid phylogenomics improves resolution of phylogenetic relationship in the Cheirostylis and Goodyera clades of Goodyerinae (Orchidoideae, Orchidaceae). Mol Phylogenet Evol. 164:107269. doi:10.1016/j.ympev.2021.107269.