
Abstract
Aureoboletus raphanaceus is a member of boletoid mushroom, which is named after its distinctive radish smell. The mitochondrial genome and phylogenetic relationships with other boletes need to be investigated to gain a comprehensive understanding of it. In this study, we sequenced the mitochondrial genome of A. raphanaceus using next-generation sequencing technology and found that its mitochondrial genome is a circular DNA molecule measuring 42,157 bp. It consists of 15 core protein-coding genes, 27 transfer RNA genes, and two ribosomal RNA genes. The mitochondrial genome had a base composition of A (39.89%), C (11.06%), G (11.67%), and T (37.38%), with a GC content of 22.73%. A phylogenetic tree based on 22 mitochondrial genomes was constructed, which provided the first insights into the phylogenetic relationships of this species with related boletes.
Introduction
Aureoboletus raphanaceus Ming Zhang & T.H. Li 2019 is an extensively distributed bolete species in southern China and symbiotic with plants of the family Fagaceae. It is distinguished by its dry and yellowish-white to pinkish-white pileus covered with fibrillose to tomentose squamules, radish smell and ovoid basidiospores (Zhang et al. Citation2019). Like many other boletes, A. raphanaceus had not been fully understood in the past and was not described until 2019 (Wu et al. Citation2016; Zhang et al. Citation2019). Accordingly, the complete mitochondrial genome of it has not been sequenced yet. Mitochondria play a vital role in the growth, adaptation, and development of fungi (Ernster and Schatz Citation1981). The mitochondrial genome size differs greatly among and within fungal species, making it a dependable genetic identifier for distinguishing species. Additionally, due to their small size, conserved orthologous genes, high copy numbers, low recombination rates, and fast evolutionary rates, fungal mitochondrial genomes are valuable for studying the evolutionary history and phylogenetic relationships (Basse Citation2010; Miao et al. Citation2022). In the present study, the complete mitochondrial genome of the East Asia bolete was sequenced on an Illumina HiSeq 2500 platform, and the phylogenetic relationships with related boletes were analyzed.
Materials
The samples of A. raphanaceus were collected from Jiuling Mountain, Jiangxi Province, China (115°15′52″E, 28°58′6″N) and were deposited in the Cryptogamic Herbarium, Kunming Institute of Botany, Chinese Academy of Sciences (http://groups.kib.cas.cn/kun/) under the voucher number KUN-HKAS 105254 (contact Kuan Zhao, [email protected]). Based on its morphological characteristics and distinct smell, the collection was identified by the corresponding author. The basidiocarps of A. raphanaceus are presented in . Specific permission is not needed as no endangered or protected species were involved.
Figure 1. The basidiocarps of Aureoboletus raphanaceus collected from Jiangxi Province, China. The specimen is distinguished by its dry and yellowish-white pileus covered with fibrillose to tomentose squamules and radish smell. Photographed by Kuan Zhao.

Methods
The total DNA was extracted from the context of the basidiocarp using the CTAB method (Doyle and Doyle Citation1987) and subsequently sequenced by Sangon Biotech Co., Ltd (Shanghai, China) for clean reads. Then, they were assembled by GetOrganelle (Jin et al. Citation2020) and the fungus database (-F fungus_mt) was employed to identify, filter, and assemble target-associated reads. The annotation was completed by MITOS Web Server using the genetic code 4 (Donath et al. Citation2019). Next, the annotated protein-coding genes (PCGs) were further refined based on the open reading frame (ORF) finder from the National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/orffinder/). The annotation of genes was examined with CPGview (Liu et al. Citation2023). Intron types were verified through RNAweasel v5.2.1 (Lang et al. Citation2007). Finally, the gene map of A. raphanaceus was visualized by PMGmap (Zhang et al. Citation2023, http://www.1kmpg.cn/pmgmap). In the phylogenetic analysis, totally 22 mitochondrial genomes were downloaded from NCBI and the Joint Genome Institute (JGI, https://mycocosm.jgi.doe.gov/mycocosm/home) database, including 19 species from the family Boletaceae and three species from the family Paxillaceae (Boletales) as outgroups. Fifteen core PCGs were individually extracted and aligned using MAFFT v7.037 (Katoh et al. Citation2019). The alignments were then concatenated to create a matrix using Phyutility v2.6 (Smith and Dunn Citation2008). The final concatenated matrix was analyzed using MrBayes v3.2.6 and RAxML v8.0.0 for Bayesian inference (BI) and maximum-likelihood (ML) methods, respectively (Ronquist and Huelsenbeck Citation2003; Stamatakis Citation2006). BI analyses were conducted under default settings (Site substitution model = Gamma site model (Gamma category = 4; GTR), Chain length of MCMC = 10,000,000, Burn-in = 10%, Model = Yule model) and terminated when the average standard deviation of split frequencies dropped below 0.01. In ML analyses, bootstrap (BS) values were assessed using the ultrafast BS approach under GTR + G model with 1000 replicates. Finally, the outcome file was visualized by MEGA7 (Kumar et al. Citation2016).
Results
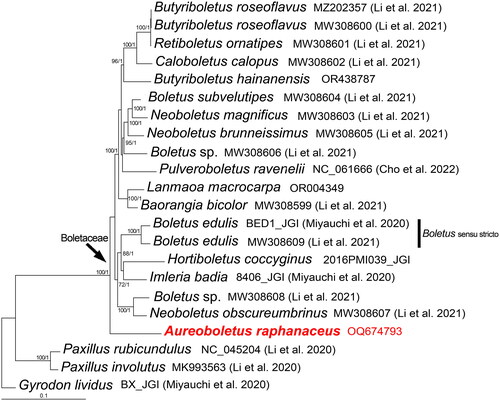
The complete mitochondrial genome sequence of A. raphanaceus (GenBank accession no. OQ674793) is shown to be 42,157 bp in length and a ×2156.84 mean coverage of trimmed sequencing data. The size of the newly sequenced species is close to that of other boletes. The gene map of A. raphanaceus is displayed in . The complete mitochondrial genome comprises 15 core PCGs (atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, and rps3), 27 transfer RNA genes and two ribosomal RNA genes. Introns, cis-splicing, and trans-splicing genes were not found in all the annotated genes. The mitochondrial genome has a base composition of A (39.89%), C (11.06%), G (11.67%), and T (37.38%), with a GC content of 22.73%. The start codon of all the 15 PCGs is ATG and the termination codon for 14 PCGs is TAA. There is one exception for atp8, where the stop codon is TAG. The phylogenetic result found that A. raphanaceus is a basal taxonomic unit in the family Boletaceae, as shown in . This finding will contribute valuable information for the further studies on the phylogeny and evolution of the genus Aureoboletus and related taxa.
Figure 2. The mitochondrial genome map of Aureoboletus raphanaceus. Genes shown outside and inside the outer circle are transcribed in counterclockwise and clockwise directions, respectively. The inner circles represent the genome scale, GC content and distributions of short tandem repeats, long tandem repeats and the dispersed repeats, respectively. The colored parabola in the center circle represents the dispersed repeats.
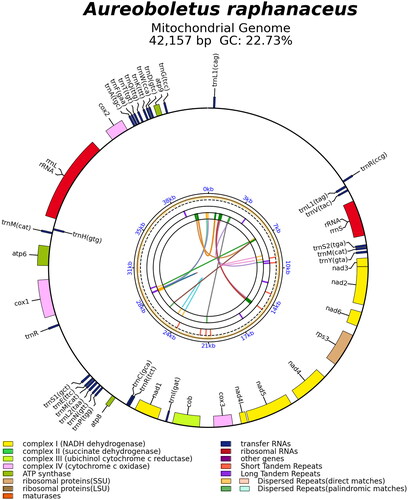
Figure 3. Phylogenetic tree of Aureoboletus raphanaceus and related taxa based on Bayesian inference (BI) and maximum-likelihood (ML) analyses of 15 core protein coding genes. The GenBank accession number from NCBI or the information of voucher specimen from JGI, along with the corresponding references (if any), are provided after the species names. The newly sequenced mitogenome is marked in red. Numbers near the nodes indicate bootstrap support values (>50%) and posterior probabilities (>0.95). The scale bar refers to 0.1 nucleotide substitutions per character.
Discussion and conclusions
In this study, the whole mitochondrial genome of A. raphanaceus was reported, which is the first species to be sequenced for mitochondrial genome from the genus. The phylogenetic analysis enriched and supplemented the previous mitochondrial genome database. As mitochondrial genome can provide more genetic information than most of the other molecular markers in phylogenetic studies, it becomes a vital potential option to discover the phylogenetic relationship of fungi (Li et al. Citation2021, Citation2023; Cho et al. Citation2022). A significant variation in length of mitochondrial genomes among different species in the Boletaceae family had been observed, which is mainly caused by differences in the intronic region (Li et al. Citation2021). The sizes of known mitochondrial genomes in boletes vary from 32,883 bp to 48,298 bp. Our recently sequenced A. raphanaceus falls in the median range with a length of 42,157 bp. In the previous study, only three out of 11 mitochondrial genomes included a single intron, the rest lacked introns. In line with this, we found no introns in the A. raphanaceus mitochondrial genome. Furthermore, this research shed light on the phylogeny of the family Boletaceae. Nonetheless, we discovered certain Chinese species identified as ‘Boletus’ that were grouped with other clades within the Boletaceae family instead of the actual Boletus senso stricto clade. This could be due to the inadequate number of mitochondrial genome data of Boletaceae currently for a comprehensive analysis of their phylogenetic relationships.
Ethical approval
No ethical issues were involved in this study. The collection of the mushroom was legal and reasonable. Information of the voucher specimen and who identified it were included in the manuscript.
Author contributions
XHM collected the samples and drafted the manuscript. XXL and YTZ analyzed the data and designed the figures. KZ designed the study, revised the manuscript, and approved the final version for publication. All authors discussed and critically revised the results and contributed to the final version of the manuscript.
Supplemental Material
Download TIFF Image (30.9 MB)Disclosure statement
The authors have declared that no competing interests exist.
Data availability statement
The mitochondrial genome data are available with the accession number of OQ674793 in the GenBank of NCBI (https://www.ncbi.nlm.nih.gov/). The associated Bioproject, SRA, and Biosample numbers are PRJNA923718, SRR23934031, and SAMN33841782, respectively.
Additional information
Funding
References
- Basse C-W. 2010. Mitochondrial inheritance in fungi. Curr Opin Microbiol. 13(6):712–719. doi: 10.1016/j.mib.2010.09.003.
- Cho S-E, Kwag Y-N, Han S-K, Lee D-H, Kim C-S. 2022. Complete mitochondrial genome sequence of Pulveroboletus ravenelii (Boletales, Basidiomycota). Mitochondrial DNA B Resour. 7(9):1581–1582. doi: 10.1080/23802359.2022.2110006.
- Donath A, Jühling F, Al-Arab M, Bernhart SH, Reinhardt F, Stadler PF, Middendorf M, Bernt M. 2019. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 47(20):10543–10552. doi: 10.1093/nar/gkz833.
- Doyle J-J, Doyle J-L. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf material. Phytochem Bull. 19:11–15. doi: 10.1016/0031-9422(80)85004-7.
- Ernster L, Schatz G. 1981. Mitochondria: a historical review. J Cell Biol. 91(3 Pt 2):227s–255s. doi: 10.1083/jcb.91.3.227s.
- Jin J-J, Yu W-B, Yang J-B, Song Y, DePamphilis C-W, Yi T-S, Li D-Z. 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):241. doi: 10.1186/s13059-020-02154-5.
- Katoh K, Rozewicki J, Yamada K-D. 2019. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 20 (4):1160–1166. doi: 10.1093/bib/bbx108.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 35(6):1547–1549. doi: 10.1093/molbev/msy096.
- Lang B-F, Laforest M-J, Burger G. 2007. Mitochondrial introns: a critical view. Trends Genet. 23(3):119–125. doi: 10.1016/j.tig.2007.01.006.
- Li Q, Luo Y-Y, Sha A-J, Xiao W-Q, Xiong Z, Chen X-D, He J, Peng L-X, Zou L. 2023. Analysis of synonymous codon usage patterns in mitochondrial genomes of nine Amanita species. Front Microbiol. 14:1134228. doi: 10.3389/fmicb.2023.1134228.
- Li Q, Wu P, Li L-J, Feng H-Y, Tu W-Y, Bao Z-J, Xiong C, Gui M-Y, Huang W-L. 2021. The first eleven mitochondrial genomes from the ectomycorrhizal fungal genus (Boletus) reveal intron loss and gene rearrangement. Int J Biol Macromol. 172:560–572. doi: 10.1016/j.ijbiomac.2021.01.087.
- Liu S, Ni Y, Li J, Zhang X, Yang H, Chen H, Liu C. 2023. CPGView: a package for visualizing detailed chloroplast genome structures. Mol Ecol Resour. 23(3):694–704. doi: 10.1111/1755-0998.13729.
- Miao Y, Chen H, Xu W, Liu C, Huang L. 2022. Cistanche species mitogenomes suggest diversity and complexity in Lamiales-Order mitogenomes. Genes. 13(10):1791. doi: 10.3390/genes13101791.
- Ronquist F, Huelsenbeck J-P. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 19(12):1572–1574. doi: 10.1093/bioinformatics/btg180.
- Smith S-A, Dunn C-W. 2008. Phyutility: a phyloinformatics tool for trees, alignments and molecular data. Bioinformatics. 24(5):715–716. doi: 10.1093/bioinformatics/btm619.
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 22(21):2688–2690. doi: 10.1093/bioinformatics/btl446.
- Wu G, Li Y-C, Zhu X-T, Zhao K, Han L-H, Cui Y-Y, Li F, Xu J-P, Yang ZL. 2016. One hundred noteworthy boletes from China. Fungal Divers. 81(1):25–188. doi: 10.1007/s13225-016-0375-8.
- Zhang M, Li T-H, Wang C-Q, Zeng N-K, Deng W-Q. 2019. Phylogenetic overview of Aureoboletus (Boletaceae, Boletales), with descriptions of six new species from China. MycoKeys. 61:111–145. doi: 10.3897/mycokeys.61.47520.
- Zhang X-Y, Chen H-M, Ni Y, Wu B, Li J-L, Burzyński A, Liu C. 2023. Plant mitochondrial genome map (PMGmap): a software tool for comprehensive visualization of coding, non-coding and genome features of plant mitochondrial genomes. Authorea. 19 October. doi: 10.22541/au.169772240.03411454/v1.