
Abstract
Saccharum species are of great importance as fruit crops due to their economic and food value. S. fulvum is a wild relative of sugarcane that has a wide geographic distribution and is well-adapted to various environmental conditions. It exhibits high resistance to pests, diseases, drought, cold, and degraded soils, making it a valuable resource for sugarcane research. Here, we report the chloroplast genome of S. fulvum. This chloroplast genome was 141,151 bp in length with a GC content of 38.41%. The large single-copy, small single-copy, and inverted repeat regions were 83,030 bp, 12,533 bp, and 22,794 bp in length, respectively. The chloroplast genome contained 111 different genes, including 77 protein-coding genes, 4 rRNA genes, and 30 tRNA genes. Phylogenetic analysis indicated that S. fulvum was closely related to S. narenga. This study not only enriches the genome information of Saccharum, but also will be useful for the evolutionary study of the family Poaceae.
Background
Saccharum L., which belongs to the family Poaceae, is an important wild relative of sugarcane that is important for sugarcane breeding research. It is widely distributed in the Americas and travels through the Mediterranean region into areas such as India, China, Southeast Asia and New Guinea. The Saccharum, Erianthus (sect. Ripidium), Sclerostachya, and Narenga form a closely related interbreeding unit involved in the origin of modern sugarcane cultivars (Mukherjee Citation1957). Saccharum fulvum R.Br. 1810 (also called Eulalia aurea (Bory) Kunth (www.worldfloraonline.org/taxon/wfo-0000896603) or Erianthus fulvus Kunth (www.worldfloraonline.org/taxon/wfo-0000868945)) can be a useful species for sugarcane varietal improvement because of its uniqueness (Kui et al. Citation2023). It has the lowest somatic chromosome count within the Saccharum complex, has a wide geographic distribution and adaptability, and shows strong resistance to pests and diseases, drought, cold, and degraded soils (Amalraj and Balasundaram Citation2006). Introducing its gene into sugarcane is expected to improve the sugarcane resistance to pests and diseases.
In recent years, chloroplast (cp) genomes of some Saccharum species have been published, such as S. hybrid (Vidigal et al. Citation2016), S. spontaneum (Vidigal et al. Citation2016), S. officinarum (Evans and Joshi, Citation2016), S. sinense (Li et al. Citation2022), S. barberi (Li et al. Citation2022), S. narenga (also called Narenga porphyrocoma) (Dyfed Lloyd and Ben, Citation2020), S. hildebrandtii (Piot et al. Citation2018). These studies have focused on comparative genomics, species classification, and origins, elucidating phylogenetic relationships within the Saccharum complex (Asano et al. Citation2004; Xu et al. Citation2019; Dyfed Lloyd and Ben, Citation2020; Li et al. Citation2022). However, the cp genome for S. fulvum has not been reported.
Therefore, we performed genome sequencing of S. fulvum and assembled its cp genome. Comparative genomic and phylogenetic analyses were then performed with other Saccharum species. Our main objective was to characterize the cp genome of S. fulvum and to determine its phylogenetic position.
Materials and methods
Plant material, DNA extraction and sequencing
In this study, we collected dry leaves of wild S. fulvum from the Sugarcane Resource Nursery of Yunnan Agricultural University, the National Crop Germplasm Resources Platform (Sugarcane), and the National Sugarcane Germplasm Resources Nursery, China (, 102.75574°E, 25.13488°N). The sample was deposited at the herbarium of the College of Pharmaceutical Engineering, Xinyang Agriculture and Forestry University (voucher number: ZM02301, Guangbo Zhang, [email protected]). Total genomic DNA was extracted using the CTAB method (Doyle and Doyle, Citation1987). The next generation sequencing DNA library with an insert size of 300 bp was constructed and sequenced on the Illumina HiSeq 2500 platform, yielding ∼4 Gb of raw data, and low-quality sequences were removed to obtain clean data.
Figure 1. Species reference map (Fruits and leaves) of S. fulvum (Wild species; Voucher number: ZM02301; This picture was taken by Guangbo Zhang from the Yunnan Agricultural University, Yunnan Province, China; 102.75574°E, 25.13488°N). S. fulvum is a near-source wild species of sugarcane that has the advantages of early maturation, high sugar content, and drought resistance that is of great value to the genetic improvements of sugarcane varieties. Core features: Culms erect, nodes long-mustachioed, upper and lower parts of nodes pilose or white powdery. Leaf blades long linear and ciliate. Panicle. Spikelet sessile and lanceolate, with white filiform hairs. Chromosome number 2n = 20. Flowering and fruiting period August-November.

Genome assembly and annotation
The cp genome assembly from the clean data was performed using GetOrganelle v. 1.7.5 (Jin et al. Citation2020). The parameters used for the plastome were ‘-R 25 -k 21,45,65,85,105,127 -F embplant_pt’. The samtools v1.7 (Li et al. Citation2009) and bedtools v2.28 (Quinlan and Hall Citation2010) were used for depth detection. The cp genome was annotated using CPGAVAS2 (Shi et al. Citation2019), PGA (Qu et al. Citation2019) and Geneious Prime v. 2022.2.2 with a reference genome (S. spontaneum, GenBank accession number: OP235381). GB2sequin (Lehwark and Greiner, Citation2019) was then used to confirm the annotation results. CPGView (Liu et al. Citation2023) was used to check the accuracy of cis- and trans-splicing genes. The cp genome map was visualized using CPGView (Liu et al. Citation2023). Sequence hotspot analysis was performed using mVISTA (Frazer et al. Citation2004) with a reference genome (S. hildebrandtii, GenBank accession number: MF563371).
Repeat and IR boundary analysis
Simple sequence repeats (SSRs) were identified using misa v. 2.1 (Beier et al. Citation2017), including mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides with minimum numbers of 10, 5, 4, 3, 3, and 3, respectively. Additionally, REPuter (Kurtz et al. Citation2001) was used to calculate palindromic, forward, reverse, and complementary repeats with the following settings: minimum repeat size of 30 bp. Furthermore, comparisons between the IR boundaries were generated using IRscope (Amiryousefi et al. Citation2018).
Phylogenetic analysis
We phylogenetically analyzed the cp genome of S. fulvum with 15 other Poaceae species. We extracted 76 common protein-coding genes (PCGs) from the genome annotation files using PhyloSuite v. 1.2.2 (Zhang et al. Citation2020). Each PCGs was aligned using MAFFT v. 7.4 (Katoh and Standley Citation2013), and then aligned genes were concatenated. Based on the concatenation matrix, a phylogenetic tree was constructed using the maximum likelihood (ML) method implemented in IQ-TREE v. 2.1.2 (Nguyen et al. Citation2014), and the best model (TPM3 + F + R5) was inferred from ModleFinder (Kalyaanamoorthy et al. Citation2017). The bootstrap value was set to 1000. Tree visualization was performed in Figtree v. 1.4.3 (https://github.com/rambaut/figtree/releases).
Results
General features of the chloroplast genome
We analyzed the coverage depth of the cp genome and tested the annotation accuracy of some difficult genes, the results indicated that the cp genome of S. fulvum was trustworthy (Figure S1; Figure S2). The cp genome of S. fulvum had a circular quadripartite structure of 141,151 bp in length (, , GenBank number: OR268641), which consisted of a large single-copy (LSC) (83,030 bp), a small single-copy (SSC) (12,533 bp), and a pair of inverted repeats (IR) (22,794 bp). This cp genome had a total GC content of 38.41%, the GC content in the IR region (43.89%) was significantly higher than that in the LSC region (36.25%) and the SSC region (32.83%). In addition, the annotation results showed that the cp genome contained 111 different genes, including 77 PCGs, 4 ribosomal RNA genes, and 30 transfer RNA genes (Table S1). Similar to other Saccharum species, the ycf1 and ycf2 genes were missing, and eight PCGs (ndhB, rpl2, rpl23, rps7, rps12, rps15, rps19, ycf15) appeared in two copies (Table S1). The variation is mainly in the spacer region compared to closely related species (Figure S3). The SSC region was more conserved than the LSC and IR regions, which have a high number of variant regions.
Figure 2. The chloroplast genome map of S. fulvum. Genes on the inside of the circle are transcribed in a clockwise direction and genes on the outside of the circle are transcribed in a counter-clockwise direction.
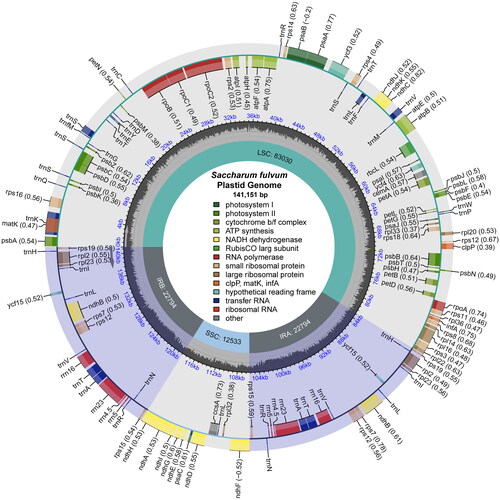
Table 1. Summary of the chloroplast genomes of S. fulvum and S. narenga species.
Repeats and IR boundaries analysis
We identified 41 simple sequence repeats (SSRs) in the cp genome of S. fulvum, including 25 mononucleotides, 5 dinucleotides, one trinucleotide, 9 tetranucleotides, and one pentanucleotide (). The majority of the SSRs were mononucleotides accounting for 60.97% of the total. The SSRs were the most abundant in the LSC region and were mainly concentrated in the non-coding regions (; Table S2). In addition, we identified 50 long repeats, including 35 forward repeats, 14 palindromic repeats, and one reverse repeat (, Table S3), which were mainly located in the LSC region, with a few presents in the IR region, and none in the SSC region (Table S3). The cp boundary genes were essentially similar in Saccharum species (Figure S4), with both the ndhH and ndhF genes located on the JSA (SSC/IRa) and JSB (SSC/IRb) boundaries. Both rpl22 and psbA genes were in the LSC region, and rps15 and rps19 genes in the IR region. However, the rps15 genes in S. hildebrandtii and S. perrieri had a shorter distance from SSC/IRb than in other Saccharum species. Compared to each other, the boundary genes of S. fulvum were essentially identical to those of other species.
Phylogenetic analysis
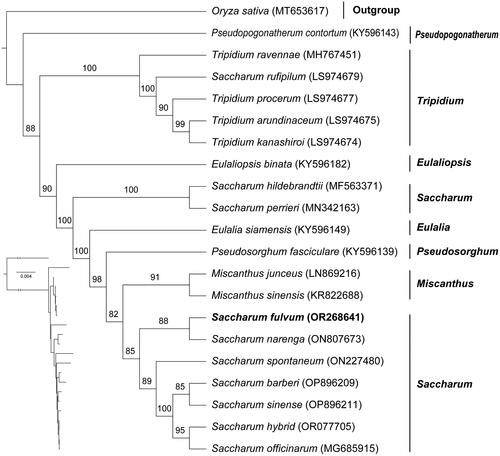
To clarify the phylogenetic position of S. fulvum, we performed a phylogenetic analysis. The phylogenetic tree showed that our phylogenetic results were generally consistent with previous studies, with most nodes having high support (). The phylogenetic analysis showed that Saccharum was not a monophyletic group. Additionally, it revealed that S. fulvum was more closely related to S. narenga than to traditional sugarcane (S. officinarum).
Figure 3. Phylogenetic tree based on the concatenated sequences of 76 protein-coding genes in 21 species by maximum-likelihood (ML). Values split by backslashes above branches represent ML bootstraps. The best-fit model was TPM3 + F + R5. Branch supports were tested using ultrafast bootstrap with 1000 replicates. Saccharum fulvum (OR268641) was marked in bold. The following sequences were used: Oryza sativa (voucher: HSAGSDYD1802) (MT653617) (Fan et al. Citation2020), Pseudopogonatherum contortum (KY596143), Tripidium ravennae (MH767451) (Lloyd Evans et al. Citation2019), Saccharum rufipilum (LS974679 (Lloyd Evans et al. Citation2019), Tripidium procerum (LS974677) (Lloyd Evans et al. Citation2019), Tripidium arundinaceum (LS974675) (Lloyd Evans et al. Citation2019), Tripidium kanashiroi (LS974674) (Lloyd Evans et al. Citation2019), Eulaliopsis binata (KY596182), Saccharum hildebrandtii (MF563371) (Piot et al. Citation2018), Saccharum perrieri (MN342163), Eulalia siamensis (KY596149), Pseudosorghum fasciculare (KY596139), Miscanthus junceus (LN869216), Miscanthus sinensis (KR822688), Saccharum narenga (ON807673) (Dyfed Lloyd and Ben, Citation2020), Saccharum spontaneum (ON227480) (Evans and Joshi, Citation2016), Saccharum barberi (OP896209) (Li et al. Citation2022), Saccharum sinense (OP896211) (Li et al. Citation2022), Saccharum hybrid (OR077705) (Li et al. Citation2022), Saccharum officinarum (MG685915) (Evans and Joshi, Citation2016).
Discussion and conclusion
Chloroplast genomes are widely used in phylogeny (Li et al. Citation2021). However, the cp genome of S. fulvum has not been previously reported. In this study, we presented the cp genome of S. fulvum, which was 141,151 bp in length and encoded a total of 111 genes. The gene order and GC content of this cp genome were similar to those of previously published Saccharum species (Dyfed Lloyd and Ben Citation2020; Li et al. Citation2022). Unlike most angiosperms, the cp genome of Saccharum species generally lacked the ycf1 and ycf2 genes, possibly due to adaptive evolution. Previous studies had not investigated the phylogeny of S. fulvum, and its exact position in the phylogenetic tree was still unclear (Asano et al. Citation2004; Evans and Joshi Citation2016; Xu et al. Citation2019; Dyfed Lloyd and Ben Citation2020; Li et al. Citation2022). Our phylogenetic results strongly supported that S. fulvum within the Saccharum branch and its close relationship with S. narenga. These findings suggested that these cp genomes could provide valuable insights into the interspecific relationships within Saccharum. However, it is important to consider the chloroplast maternal inheritance, which limits the accuracy of phylogenetic analysis (Krawczyk et al. Citation2018). To obtain more precise phylogenetic relationships, it is necessary to integrate the analysis of nuclear and organelle genomes (Górniak et al. Citation2010). Additionally, future research should include genomic analyses of other Saccharum species to further explore the complexity of Saccharum. This study not only enhances the genomic information of Saccharum but also serves as a basis for understanding the evolution of Poaceae species.
Ethical approval
No permission from the People’s Republic of China government was required to collect these plants. The author has read the manuscript and has approved this submission. Ethical statement is not applicable. The research on plants used in this study, including the collection of plant material has been carried out in accordance with guidelines provided by our institution. Field studies in our manuscript have complied with local legislation and appropriate permissions/license were granted while taking samples from a preserved/protected land.
Consent form
The authors complied with relevant institutional (Xinyang Agriculture and Forestry University), national (the People’s Republic of China), and international guidelines (IUCN) and legislation for the plant study.
Authors’ contributions
Guangbo Zhang directed the study and designed the experiments. Jing Li and Xiuqing Liu performed the material sampling. Siying Zhang performed the data processing and drafted the manuscript. All authors approved the final draft.
Supplemental Material
Download MS Excel (15.2 KB)Supplemental Material
Download MS Excel (14.6 KB)Supplemental Material
Download MS Excel (12 KB)Supplemental Material
Download JPEG Image (821 KB){kind=link}
Supplemental Material
Download JPEG Image (1.4 MB){kind=link}
Supplemental Material
Download JPEG Image (443.5 KB){kind=link}
Supplemental Material
Download JPEG Image (1.1 MB){kind=link}
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The genome sequence data that support the findings of this study are available in GenBank of NCBI (http://www.ncbi.nlm.nih.gov/) under the accession no. OR268641. The associated BioProject, BioSample, and SRA numbers are PRJNA1005584, SAMN36990507, SRR25653907, respectively.
Additional information
Funding
References
- Amalraj VA, Balasundaram NJ. 2006. On the taxonomy of the members of ‘Saccharum complex’. Genet Resour Crop Evol. 53(1):35–41. doi:10.1007/s10722-004-0581-1.
- Amiryousefi A, Hyvönen J, Poczai P. 2018. IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics. 34(17):3030–3031. doi:10.1093/bioinformatics/bty220.
- Asano T, Tsudzuki T, Takahashi S, Shimada H, Kadowaki K-i. 2004. Complete nucleotide sequence of the sugarcane (Saccharum officinarum) chloroplast genome: a comparative analysis of four monocot chloroplast genomes. DNA Res. 11(2):93–99. doi:10.1093/dnares/11.2.93.
- Beier S, Thiel T, Münch T, Scholz U, Mascher M., 2017. MISA-web: a web server for microsatellite prediction. Bioinformatics. 33(16):2583–2585. doi:10.1093/bioinformatics/btx198.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissues. Phytochem Bull. 19:11–15.
- Dyfed Lloyd E, Ben H. 2020. Complete Chloroplast Genomes of Saccharum giganteum, Saccharum longisetosum, Cleistachne sorghoides, Sarga timorense, Narenga porphyrocoma, Tripsacum dactyloides. Comparisons with ITS Phylogeny and Placement within Saccharum. bioRxiv. 12:1–44
- Evans DL, Joshi SV. 2016. Complete chloroplast genomes of Saccharum spontaneum, Saccharum officinarum and Miscanthus floridulus (Panicoideae: Andropogoneae) reveal the plastid view on sugarcane origins. Syst Biodivers. 14(6):548–571. doi:10.1080/14772000.2016.1197336.
- Fan J, Zhu WY, Li ZF, et al. 2020. Chloroplast genome sequence of a yellow colored rice (Oryza sativa L.): insight into the genome structure and phylogeny. Mitochondrial DNA Part B Resour. 5(3):3650–3652.
- Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. 2004. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32(Web Server issue):W273–279., doi:10.1093/nar/gkh458.
- Górniak M, Paun O, Chase MW. 2010. Phylogenetic relationships within Orchidaceae based on a low-copy nuclear coding gene, Xdh: congruence with organellar and nuclear ribosomal DNA results. Mol Phylogenet Evol. 56(2):784–795. doi:10.1016/j.ympev.2010.03.003.
- Jin J-J, Yu W-B, Yang J-B, Song Y, dePamphilis CW, Yi T-S, Li D-Z., 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):241. doi:10.1186/s13059-020-02154-5.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS., 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589. doi:10.1038/nmeth.4285.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780. doi:10.1093/molbev/mst010.
- Krawczyk K, Nobis M, Myszczyński K, Klichowska E, Sawicki J., 2018. Plastid super-barcodes as a tool for species discrimination in feather grasses (Poaceae: Stipa). Sci Rep. 8(1):1924. doi:10.1038/s41598-018-20399-w.
- Kui L, Majeed A, Wang X, Yang Z, Chen J, He L, Di Y, Li X, Qian Z, Jiao Y, et al. 2023. A chromosome-level genome assembly for Erianthus fulvus provides insights into its biofuel potential and facilitates breeding for improvement of sugarcane. Plant Commun. 4(4):100562. doi:10.1016/j.xplc.2023.100562.
- Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R., 2001. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29(22):4633–4642. doi:10.1093/nar/29.22.4633.
- Lehwark P, Greiner S. 2019. GB2sequin - A file converter preparing custom GenBank files for database submission. Genomics. 111(4):759–761. doi:10.1016/j.ygeno.2018.05.003.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R., 2009. The sequence alignment/map format and SAMtools. Bioinformatics. 25(16):2078–2079. doi:10.1093/bioinformatics/btp352.
- Li J, Tang J, Zeng S, Han F, Yuan J, Yu J., 2021. Comparative plastid genomics of four Pilea (Urticaceae) species: insight into interspecific plastid genome diversity in Pilea. BMC Plant Biol. 21(1):25. doi:10.1186/s12870-020-02793-7.
- Li S, Duan W, Zhao J, Jing Y, Feng M, Kuang B, Wei N, Chen B, Yang X. 2022. Comparative analysis of chloroplast genome in Saccharum spp. and related members of Saccharum complex. Int J Mol Sci. 23(14):7661. doi:10.3390/ijms23147661.
- Liu S, Ni Y, Li J, Zhang X, Yang H, Chen H, Liu C., 2023. CPGView: a package for visualizing detailed chloroplast genome structures. Mol Ecol Resour. 23(3):694–704. doi:10.1111/1755-0998.13729.
- Lloyd Evans D, Joshi SV, Wang J. 2019. Whole chloroplast genome and gene locus phylogenies reveal the taxonomic placement and relationship of Tripidium (Panicoideae: Andropogoneae) to sugarcane. BMC Evol Biol. 19(1):33. doi:10.1186/s12862-019-1356-9.
- Mukherjee SK. 1957. Origin and distribution of Saccharum. Botanical Gazette. 119(1):55–61. doi:10.1086/335962.
- Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ., 2014. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274. doi:10.1093/molbev/msu300.
- Piot A, Hackel J, Christin P-A, Besnard G., 2018. One-third of the plastid genes evolved under positive selection in PACMAD grasses. Planta. 247(1):255–266. doi:10.1007/s00425-017-2781-x.
- Qu X-J, Moore MJ, Li D-Z, Yi T-S. 2019. PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods. 15(1):50. doi:10.1186/s13007-019-0435-7.
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26(6):841–842. doi:10.1093/bioinformatics/btq033.
- Shi L, Chen H, Jiang M, Wang L, Wu X, Huang L, Liu C., 2019. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47(W1):W65–W73. doi:10.1093/nar/gkz345.
- Vidigal PMP, Coelho ASG, Novaes E, Barbosa MHP, Peternelli LA., 2016. Complete chloroplast genome sequence and annotation of the Saccharum Hybrid Cultivar RB867515. Genome Announc. 4(5):e01157-01116. doi:10.1128/genomeA.01157-16.
- Xu F, He L, Gao S, Su Y, Li F, Xu L. 2019. Comparative analysis of two sugarcane ancestors Saccharum officinarum and S. spontaneum based on complete chloroplast genome sequences and photosynthetic ability in cold stress. Int J Mol Sci. 20(15):3828. doi:10.3390/ijms20153828.
- Zhang D, Gao F, Jakovlić I, Zou H, Zhang J, Li WX, Wang GT., 2020. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour. 20(1):348–355. doi:10.1111/1755-0998.13096.