
Abstract
The brown-eared bulbul (Hypsipetes amaurotis) is a medium-sized songbird native to East Asia and characterized by its prominent reddish-brown ear-coverts. Previous studies on it have primarily been from the taxonomic and morphological aspects, with limited research in the realm of molecular biology. In this study, we sequenced and annotated the complete mitochondrial genome of H. amaurotis, which was the first reported complete mitogenome of the genus Hypsipetes. The mitogenome of H. amaurotis is 17,871 bp in length and was predicted to encode 37 typical mitochondrial genes, including 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNAs), two ribosomal RNA genes (rRNAs). Specifically, this mitogenome contains two D-loop control regions that are of similar length and sequencing pattern. A total of 8 Pycnonotidae and six outgroup taxa were used to determine the phylogenetic placement with two methods: Maximum Likelihood Approximation (IQ-TREE) and Bayesian inference (MrBayes). Our findings reveal that H. amaurotis is phylogenetically closely related to Ixos mcclellandii. The outcomes are generally consistent with the phylogenetic trees constructed in previous studies. The data gathered from this research provides valuable insights for future genomic investigations into the evolution, ecology, and conservation of this species.
1. Introduction
The brown-eared bulbul (Hypsipetes amaurotis; Temminck et al. Citation1838) is a medium-sized songbird, measuring 27–29 cm in length, with prominent reddish-brown ear-coverts. It is distributed in Japan, the Korean Peninsula, the Philippines, and China. This bulbul is typically seen either solitary or in small groups within the upper canopy or along the edges of forests (Zheng 1985). The varied diet of H. amaurotis consists of both plant and animal matter. During summer, its primary food source is insects, but it shifts toward fruits and seeds during fall and winter (Zhao Citation2001).
As for naming, although commonly known as H. amaurotis, its taxonomic classification has undergone multiple revisions over time. It was initially classified under the genus Turdus (Temminck et al. Citation1838), subsequently switched to Ixos (Sibley and Monroe Citation1990), and later, Microscelis (Hoyo and Jutglar Citation1992). It was not until 2010 that it was finally classified to its current designation of the genus Hypsipetes of the family Pycnonotidae (Gill et al. Citation2023). Previous research was predominantly on taxonomy and morphology, so a molecular perspective is seriously needed and can be revealing.
Our study presents the first complete sequence of H. amaurotis mitogenome. Based on it, a comparison was made with related species in terms of mitochondrial structure and gene rearrangement. Furthermore, the phylogenetic relationships between H. amaurotis and other species within the Pycnonotidae family were explored through analysis of the comprehensive set of mitochondrial genes. The newly obtained complete mitogenome sequence is valuable data for future research on this understudied genus.
2. Materials and methods
The bird specimens used in this study were captured with mist nets on April 17, 2023, from the Xianrendong National Nature Reserve, Liaoning Province, China (39°59′16″N, 122°57′42″E) (). Muscle tissue samples for the experiment were taken from the chest and stored at a temperature of −80 °C in a 100% ethanol solution before DNA extraction. A specimen was deposited at School of Life Sciences, Liaoning University (Prof. Dongmei Wan, E-mail: [email protected]) under the voucher number hyps_ama_i1_t1.
Figure 1. Photograph of H. amaurotis. Photographed by Chaofan Feng, and copyright license agreements were obtained.

Total genomic DNA was extracted using TIANamp Genomic DNA Kit (DP304, TIANGEN, Beijing, China) following the manufacturer’s instructions. The MGI DNB-seq-T7 platforms with 300 cycles of paired-end sequencing (150 bp reads) were utilized at Sangon Biotech (Shanghai) Co., Ltd., Shanghai, China.
Firstly, fastp v0.20.0 (S. Chen et al. Citation2018) was employed to perform quality control. Then, read assembly was run using getorganelle v1.7.7.0 (Jin et al. Citation2020) employing SPAdes v3.15.5 (Bankevich et al. Citation2012) with the maximum value of k-mer set at 141. Subsequently, gene annotation was initially performed using MitoZ v3.6 (Meng et al. Citation2019) and MitoAnnotator online service v3.92 (Zhu et al. Citation2023) and confirmed manually through comparisons with homologous genes previously published of Pycnonotidae species.
A total of 7 Pycnonotidae mitogenome sequences were collected from the NCBI Nucleotide database (https://www.ncbi.nlm.nih.gov/nucleotide/) as references, which covered all Pycnonotidae species whose complete sequence is known. Additionally, 6 mitogenomes from closely related families were downloaded and served as outgroups (Table S1).
The alignment process was performed independently for each gene using mafft v7.520 (Yamada et al. Citation2016). A partitioning scheme containing 5 sections was created based on tRNA genes, rRNA genes, and 3 codon positions of PCGs. Phylogenetic trees were constructed using both Bayesian inference (BI) and maximum likelihood (ML) methods. ML analysis was performed using IQ-TREE v2.2.2.3 (Minh et al. Citation2020) through standard model selection and conducted on 1000 ultrafast bootstrap replicates. The best-fit models were chosen by embedded ModelFinder (Kalyaanamoorthy et al. Citation2017) according to the Bayesian information criterion (BIC). The BI method was performed using MrBayes v3.2.7 (Ronquist et al. Citation2012) with 4 simultaneous Markov chain Monte Carlo (MCMC) chains running for 1,000,000 generations and sampling every 1,000 generations with a burn-in of 25%.
3. Results
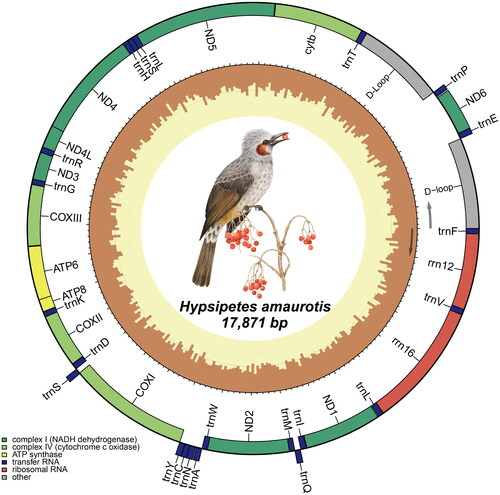
The complete mitogenome of H. amaurotis was assembled from 1.86 Gb worth of raw data with an average depth of 71,105.44 x (Figure S1), which covered a total of 17,871 base pairs and contained 13 protein-coding genes (PGCs), 22 transfer RNA genes(tRNAs), 2 ribosomal RNA genes(rRNAs), and 2 non-coding control regions (, Table S2). The order and orientation of the mitochondrial genes were consistent with other members of the Pycnonotidae family (Figure S2). All of the PCGs started with ATG codon except for COXI, which started with GTG codon. Three PCGs (ND2, COXIII, ND4) had incomplete stop codons, while the other 10 PCGs ended with complete vertebrate mitochondrial stop codons. The tRNAs had a length range of 58 to 85 bp and could fold into a cloverleaf secondary structure, except for tRNA-Ser (GCT) which had an incomplete secondary structure due to the absence of the D-arm. Additionally, the two control regions exhibited a similarity of 95.39%, representing approximately 1000 bp of identical sequences, and both could be divided into three domains (Figure S3).
Figure 2. Graphical map of the complete mitogenome of H. amaurotis. Genes encoded by the heavy strand are shown inside the circle, whereas those encoded by the light strand are shown outside the circle.
A total of 8 Pycnonotidae and 6 outgroup mitogenomes were used to reconstruct phylogenetic relationships by the BI and ML methods. Both methods generated similar tree topologies except for the position of Rubigula melanicterus, which was pruned in the BI tree due to the low probability (). The phylogenetic tree indicated that H. amaurotis belongs to the family Pycnonotidae and is closely related to mountain bulbul (I. mcclellandii).
Figure 3. Mitochondrial phylogenetic relationships among family pycnonotidae (see Table S1). Bayesian inference and maximum likelihood analysis supported the same topological structure. Values at nodes are Bayesian posterior probabilities (BPPs) and ML bootstrap probabilities (BSPs). An asterisk * indicates a BPP of 1.0 and 100% BSP. Node with a bootstrap support below 50% is collapsed. The following sequences were used: Hypsipetes amaurotis OR473627, Ixos mcclellandii NC_039396 (Chen et al. Citation2018), Spizixos semitorques NC_029321 (Ren et al. Citation2016), Rubigula melanicterus NC_024730 (Ren et al. Citation2016), Pycnonotus jocosus.
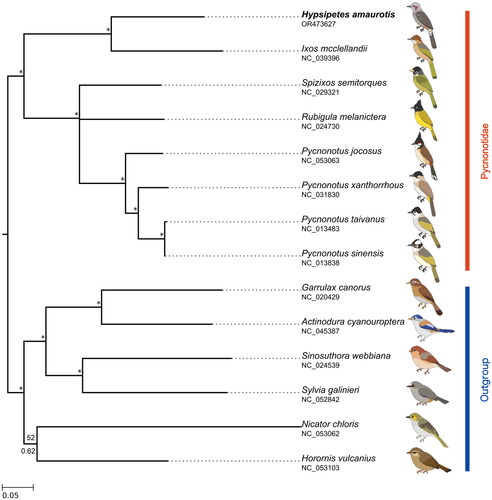
NC_053063 (Feng et al. Citation2020), Pycnonotus xanthorrhous NC_031830 (Wen and Liao Citation2016), Pycnonotus taivanus NC_013483 (Chang et al. Citation2010), Pycnonotus sinensis NC_013838 (Chang et al. Citation2010), Garrulax canorus NC_020429 (D. S. Chen et al. Citation2015), Actinodura cyanouroptera NC_045387 (He et al. Citation2019), Sinosuthora webbiana NC_024539 (Zhang et al. Citation2015), Sylvia galinieri NC_052842 (Manthey et al. Citation2022), Nicator chloris NC_053062 (Feng et al. Citation2020), Horornis vulcanius NC_053103 (Feng et al. Citation2020).
4. Discussion and conclusion
In the present study, we assembled and analyzed the complete mitogenome of H. amaurotis. The mitogenome length (17,871bp) of H. amaurotis was close to that of Ixos mcclellandii (17,838bp) and some 800 bp longer than those of other Pycnonotidae species (Table S1). On the other hand, the gene order of mitogenome is consistent across the Pycnonotidae family (Figure S3). Based on this, the observed size variations might be attributed to differences in intragenic spacer sizes, with the control region playing a prominent role.
In the mitogenomes of all available species within the family, two control regions were observed. But they fell into two scenarios: the majority of species exhibited two distinct control regions, whereas H. amaurotis and I. mcclellandii possessed two highly similar complete control regions. This completeness may have evolutionary advantages, and therefore, in some cases, are retained, or not completely degraded (Eberhard et al. Citation2001).
The observed phylogenetic tree topology in this study is mostly consistent with previous investigations on Pycnonotidae (Oliveros and Moyle Citation2010; Shakya and Sheldon Citation2017), except for the unresolved placement of Rubigula melanicterus. This is due to low support values and a relatively limited number of species. The close relationship of H. amaurotis and I. mcclellandii in the phylogenetic tree, combined with the similar scenarios observed in their control regions, suggests that they share a recent common ancestor.
In conclusion, our results provide the first complete mitogenome of the H. amaurotis and even for the genus Hypsipetes. Due to the possession of two highly similar complete control regions, the mitogenome of H. amaurotis was 800 bp longer than those of most other Pycnonotidae species. Our results were generally consistent with previous studies on phylogenetic trees involving H. amaurotis. The information obtained holds valuable insights for understanding H. amaurotis and future genomic research on this species.
Ethical approval
The material involved in the article does not involve ethical conflicts. This study was permitted by the School of Life Sciences, Liaoning University, Liaoning, China. All collection and sequencing work were strictly executed under local legislation and related laboratory regulations to protect wild resources.
Authors’ contributions
YZ and DW contributed to the study conception and design. Material preparation, data collection, and analysis were performed by YL, YZ, and BR. The first draft of the manuscript was written by YL and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Supplemental Material
Download MS Word (2.3 MB)Disclosure statement
The authors report no potential conflict of interest.
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI (https://www.ncbi.nlm.nih.gov/) under the accession no. OR473627. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA1008370, SRR25736572, and SAMN37117600 respectively.
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477. doi: 10.1089/cmb.2012.0021.
- Baolai Z. 1985. Fauna Sinica. Vol. Passeriformes (Eurylaimidae lrenidae). Vol. 11. Aves. Science Press.
- Chang H-W, Chou Y-C, Su Y-F, Cheng C-A, Yao C-T, Tsai C-L, Lee H-C, Wen C-H, Cheng C-C. 2010. Molecular phylogeny of the Pycnonotus sinensis and Pycnonotus taivanus in Taiwan based on sequence variations of nuclear CHD and mitochondrial cytochrome b genes. Biochem Syst Ecol. 38(2):195–201. doi: 10.1016/j.bse.2009.12.016.
- Chen DS, Qian CJ, Ren QQ, Wang P, Yuan J, Jiang L, Bi D, Zhang Q, Wang Y, Kan XZ. 2015. Complete mitochondrial genome of the Chinese Hwamei Garrulax Canorus (Aves: passeriformes): the first representative of the Leiothrichidae family with a duplicated control region. Genet Mol Res. 14(3):8964–8976. doi: 10.4238/2015.August.7.5.
- Chen Y, Song X, Zhou C, Zhang W, Chen B, Wu S, Bisong YUE. 2018. Sequencing and analysis of the mitochondrial genome of Ixos mcclellandii. Sichuan J Zoo. 37(6):646–652.
- Chen S, Zhou Y, Chen Y, Gu J. 2018. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 34(17):i884–i890. doi: 10.1093/bioinformatics/bty560.
- Eberhard JR, Wright TF, Bermingham E. 2001. Duplication and concerted evolution of the mitochondrial control region in the parrot genus Amazona. Mol Biol Evol. 18(7):1330–1342. doi: 10.1093/oxfordjournals.molbev.a003917.
- Feng S, Stiller J, Deng Y, Armstrong J, Fang Q, Reeve AH, Xie D, Chen G, Guo C, Faircloth BC, et al. 2020. Dense sampling of bird diversity increases power of comparative genomics. Nature. 587(7833):252–257. doi: 10.1038/s41586-020-2873-9.
- Gill F, Donsker D, Rasmussen P. 2023. “IOC World Bird List 13.1.” doi: 10.14344/IOC.ML.13.1.
- He W, Xu H, Li D, Xie M, Zhang M, Ni Q, Yao Y. 2019. The complete mitochondrial genome of Blue-Winged Minla (Minla Cyanouroptera) and its phylogenetic analysis. Mitochondrial DNA B Resour. 4(2):2784–2785. doi: 10.1080/23802359.2019.1659109.
- Hoyo J, Jutglar F. 1992. Handbook of the Birds of the World. Lynz Edicions. https://books.google.com.sg/books?id=CMLNzAEACAAJ.
- Jin J-J, Yu W-B, Yang J-B, Song Y, dePamphilis CW, Yi T-S, Li D-Z. 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):241. doi: 10.1186/s13059-020-02154-5.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589. doi: 10.1038/nmeth.4285.
- Manthey JD, Bourgeois Y, Meheretu Y, Boissinot S. 2022. Varied diversification patterns and distinct demographic trajectories in Ethiopian montane forest bird (Aves: passeriformes) populations separated by the Great Rift Valley. Mol Ecol. 31(9):2664–2678. doi: 10.1111/mec.16417.
- Meng G, Li Y, Yang C, Liu S. 2019. MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 47(11):e63–e63–e63. doi: 10.1093/nar/gkz173.
- Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, Lanfear R. 2020. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 37(5):1530–1534. doi: 10.1093/molbev/msaa015.
- Oliveros CH, Moyle RG. 2010. Origin and diversification of Philippine bulbuls. Mol Phylogenet Evol. 54(3):822–832. doi: 10.1016/j.ympev.2009.12.001.
- Ren Q, Qian C, Yuan J, Jiang L, Wang P, Kan X. 2016. Characterization of the complete mitochondrial genome of collared Finchbill, Spizixos Semitorques (Passeriformes: pycnonotidae). Mitochondrial DNA A DNA Mapp Seq Anal. 27(1):700–702. doi: 10.3109/19401736.2014.913150.
- Ren Q, Qian C, Yuan J, Li X, Yang J, Wang P, Jiang L, Zhang Q, Wang Y, Kan X. 2016. Complete mitochondrial genome of the black-capped Bulbul, Pycnonotus Melanicterus (Passeriformes: pycnonotidae). Mitochondrial DNA A DNA Mapp Seq Anal. 27(2):1378–1380. doi: 10.3109/19401736.2014.947589.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61(3):539–542. doi: 10.1093/sysbio/sys029.
- Shakya SB, Sheldon FH. 2017. The phylogeny of the world’s Bulbuls (Pycnonotidae) inferred using a supermatrix approach. Ibis. 159(3):498–509. doi: 10.1111/ibi.12464.
- Sibley CG, Monroe BL. 1990. Distribution and taxonomy of birds of the world. Yale University Press. https://books.google.com.sg/books?id=Wk-vyrNVAccC.
- Temminck CJ, Buffon Comte de GLL, Huet N, de Chartrouse Baron GMJML, Prêtre JG. 1838. (CoenraadJacob) Nouveau Recueil de Planches Coloriées d’oiseaux : Pour Servir de Suite et de Complément Aux Planches Enluminées de Buffon, Édition in-Folio et in-4° de l’Imprimerie Royale, 1770. Vol. 1 (1838). A Strasbourgh: Chez Legras Imbert et Comp. https://www.biodiversitylibrary.org/item/109466.
- Wen L, Liao F. 2016. Complete mitochondrial genome of Pycnonotus xanthorrhous (Passeriformes, Pycnonotidae) and phylogenetic consideration. Biochem Syst Ecol. 69(December):83–90. doi: 10.1016/j.bse.2016.08.009.
- Yamada KD, Tomii K, Katoh K. 2016. Application of the MAFFT sequence alignment program to large data—reexamination of the usefulness of chained guide trees. Bioinformatics. 32(21):3246–3251. doi: 10.1093/bioinformatics/btw412.
- Zhang H, Li Y, Wu X, Xue H, Yan P, Wu X-B. 2015. The complete mitochondrial genome of Paradoxornis webbianus (Passeriformes, Muscicapidae). Mitochondrial DNA. 26(6):879–880. doi: 10.3109/19401736.2013.861440.
- Zhao Z-j. 2001. A handbook of the birds of China. Jilin Science and Technology Press.
- Zhu T, Sato Y, Sado T, Miya M, Iwasaki W. 2023. MitoFish, MitoAnnotator, and MiFish Pipeline: updates in 10 Years. Mol Biol Evol. 40(3):msad035. doi: 10.1093/molbev/msad035.