
Abstract
Cyanopterus ninghais is an important gregarious ectoparasitoid during the larval stage of Monochamus alternatus, a key vector for pine wilt disease in Asia. In this study, the complete mitochondrial genome of C. ninghais was sequenced and analyzed. The mitochondrial genome of C. ninghais is 15,386 bp in length, comprising 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNAs), and 2 ribosomal RNA genes (rRNAs). The nucleotide composition is 41.32% A, 8.29% G, 6.06% C, and 44.33% T. Phylogenetic trees of Braconidae were constructed using 13 PCG sequences via Bayesian inference (BI) and maximum likelihood (ML) analyses to determine their phylogenetic position. Both ML and BI analyses revealed that C. ninghais is closely related to Euurobracon yokahamae, Virgulibracon endoxylaphagus, and Habrobracon hebetor.
Keywords:
Introduction
Cyanopterus (Bracomorpha) ninghais (Wang et al. Citation2009) (Hymenoptera: Braconidae) is a gregarious ectoparasitic wasp that targets the third to fifth instar larval stage of the Japanese pine sawyer beetle, Monochamus alternatus (Hope, 1842) (Coleoptera: Cerambycidae). This beetle is the primary vector insect responsible for the spread of pine wilt disease, a significant concern in Asia (Wang et al. Citation2009; Li et al. Citation2021; Xu et al. Citation2023a). Cyanopterus ninghais shows a strong host specificity for M. alternatus, which indicates a significant potential for its use as a biological control agent. Despite its ecological significance, research on the Cyanopterus genus is currently limited to morphological classification, and the complete mitochondrial genome sequence of C. ninghais has not been released in public databases. The absence of genetic information restricts our exploration of the phylogenetic relationships within this genus, thus impeding our comprehension of the evolution of Braconidae. Mitochondrial genomes have been extensively utilized in phylogenetic studies because of their maternal inheritance, relatively fast evolutionary rate, and limited recombination. Using mitochondrial genomes for phylogenetic construction is a common approach and has been successfully applied in numerous studies. For instance, Carter proposed detailed phylogenetic hypotheses for 10 species of the genus Hirundo using complete mitochondrial genomes (Carter et al. Citation2020). In this study, we determined the whole mitochondrial genome of C. ninghais for the first time and analyzed the phylogenetic relationships of C. ninghais with other Braconid species, which provides valuable genetic resources for further studies on the phylogeny of the Cyanopterus genus.
Materials
The sample of C. ninghais was collected from Zunyi, Guizhou Province, China (27.59 N, 106.83 E) in June 2022 (). After the collection, the voucher specimen No. ZYgqh_0001 (contact Xiaoyi Wang, [email protected]) was stored in 100% ethanol and is presently located in the Entomological Museum of the Chinese Academy of Forestry. The corresponding author identified the collection based on its morphological characteristics.
Figure 1. The female specimen of Cyanopterus ninghais in dorsal view (collected from Zunyi, China, and the picture taken by Shaobo Wang in 2022).

Methods
Mitochondrial genome assembly and annotation
Total DNA was extracted from whole body using E.Z.N.A.® Tissue DNA Kit (Omega, GA, USA). To obtain a complete mitochondrial genome sequence, we used both short-read sequencing (Illumina NovaSeq 6000 platform) and long-read sequencing (PacBio Sequel platform) technologies in this study. The read coverage depth map is shown in Supplementary Figure S1. The assembly of the short clean reads using GetOrganelle v1.7.5 (Jin et al. Citation2020), followed by their alignment to the PacBio long reads using BWA v0.7.17. We then extracted the specific PacBio long reads corresponding to our target sample. To achieve a comprehensive assembly, we used SPAdes v3.14.1 to integrate the extracted PacBio long reads with the short clean reads. Sequences with sufficient coverage depth and longer assembly lengths were carefully selected as candidate sequences, and their accuracy was confirmed by alignment with the Nucleotide Sequence Database, followed by sequence joining based on overlap. To ensure data accuracy, the clean reads were resequenced to the mitochondrial genome, and base corrections were performed using Pilon v1.23. The mitogenome genome of C. ninghais was annotated using MITOS (Bernt et al. Citation2013), enabling the prediction of protein-coding genes (PCGs), transfer RNA (tRNA) genes, and ribosomal RNA (rRNA). Finally, the circular mitochondrial genome map of C. ninghais was drawn using CGView (Grant and Stothard Citation2008).
Phylogenetic analysis
To infer the taxonomic position of C. ninghais, a total of 26 mitochondrial genomes were downloaded from NCBI, including 26 species from the family Braconidae and one species from the family Ichneumonidae (Insecta, Hymenoptera) as outgroup. Phylogenetic analysis of taxa within the Braconidae family was conducted using PhyloSuite v1.2.2 (Zhang et al. Citation2020a). The mitochondrial genomes of 27 species were utilized for this analysis, with the 13 PCGs individually aligned using the MAFFT algorithm. Subsequently, sequence alignments were refined and trimmed using trimAL v1.2. The 13 PCGs were combined to form a unified dataset, constituting the fundamental dataset underpinning our phylogenetic analysis. Maximum likelihood (ML) phylogenies were inferred by employing IQ-TREE (Nguyen et al. Citation2015). The partition schemes and best-fit substitution models were determined by ModelFinder, which employs the corrected Akaike information criterion. Node support was assessed through the generation of 1000 ultrafast bootstrap replicates. In parallel, Bayesian inference (BI) phylogenies were constructed using MrBayes (Ronquist et al. Citation2012). Two independent runs were conducted, each employing four Markov chain Monte Carlo chains, including one cold chain. These runs were executed for 5,000,000 generations, with sampling occurring at intervals of 1000 generations. The initial 25% of the runs were discarded as burn-in, and posterior probabilities were subsequently calculated to estimate branch support. The optimal partitioning schemes and substitution models for the BI analysis were determined using ModelFinder (Kalyaanamoorthy et al. Citation2017), which employs the corrected Bayesian information criterion.
Results
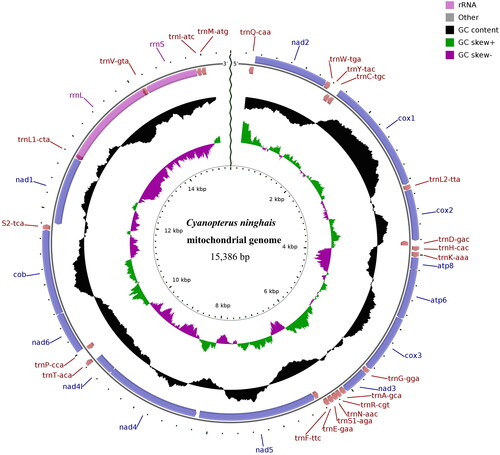
The complete mitochondrial genome of C. ninghais (GenBank accession no. OR525636) is circular and has a length of 15,386 bp. It encodes a total of 13 PCGs, 22 transfer RNA genes (tRNAs), and two ribosomal RNA genes (rRNAs). The overall base composition of mitogenome is 41.32% A, 8.29% G, 6.06% C, and 44.33% T, with a GC content of 14.36% (; Supplementary Figure S2). The cumulative length of the 13 PCGs is 11,124 bp, with PCG lengths ranging from 282 bp to 1665 bp, accounting for 72.30% of the complete genome. The cumulative length of the tRNA genes is 1469 bp, with an average gene length of 67 bp, accounting for 9.55% of the complete genome. The lengths of rRNA genes are 1364 bp (rrnL) and 750 bp (rrnS), accounting for 13.74% of the complete genome. Among the 13 PCGs, 5 PCGs start with ATT (cox2, nad1, nad2, nad3, nad4l), 4 PCGs start with ATA (atp6, atp8, nad4, nad5) and 4 PCGs start with ATG (cob, cox1, cox3, nad6). Except for nad3, which ends with TAG, the remaining 12 PCGs all end with TAA (Supplementary Table S1).
Figure 2. Mitochondrial genome map of Cyanopterus ninghais. Clockwise genes on the outer circle are on the H-strand, and counterclockwise genes on the inner circle are on the L-strand.
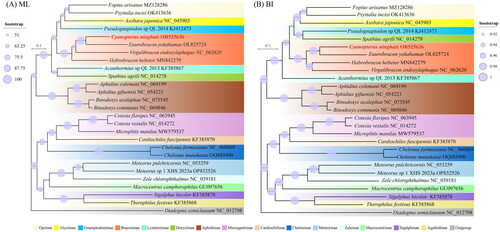
To infer the evolutionary relationship of C. ninghais, a phylogenetic analysis was performed based on the nucleotide sequences of mitochondrial genomes from 26 complete mitochondrial genomes within the family Braconidae. The topologies constructed by ML and BI analyses based on the PCGs dataset were almost identical, supporting a close phylogenetic relationship between C. ninghais and Euurobracon yokahamae (Dalla Torre, 1898), Virgulibracon endoxylaphagus (Quicke & Ingram, 1993), and Habrobracon hebetor (Say, 1836) ().
Figure 3. Phylogenetic trees of Braconidae produced from the maximum likelihood (A) and Bayesian inference (B) analyses based on 13 protein-coding genes. The bootstrap support value (A) and the posterior probabilities (B) are indicated by the size of the circle around the node. The red font represented the target species in this study. The following sequences were used: Therophilus festivus (Muesebeck, 1953) KF385868, Asobara japonica (Belokobylskij, 1998) NC_045903 (Zhang et al. Citation2020b), Aphidius gifuensis (Ashmead, 1904) NC_054223 (Feng et al. Citation2020), Aphidius colemani (Viereck, 1912) NC_069199, Binodoxys communis (Gahan, 1927) NC_069846, Binodoxys acalephae (Marshall, 1896) NC_073545 (Xu et al. Citation2023b), Virgulibracon endoxylaphagus NC_062620, Habrobracon hebetor MN842279 (Huang et al. Citation2020), Euurobracon yokohamae OL825724 (Kim et al. Citation2022), Cardiochiles fuscipennis (Viereck, 1912) KF385870, Chelonus formosanus (Sonan, 1932) NC_060869 (Yuan et al. Citation2022), Chelonus munakatae (Sonan, 1932) OQ885490, Spathius agrili (Yang, 2005) NC_014278 (Wei et al. Citation2010), Pseudognaptodon sp. QL-2014 KJ412473, Acanthormius sp. QL-2013 KF385867, Macrocentrus camphoraphilus (Viereck, 1912) GU097656 (Wei et al. Citation2010), Meteorus sp. XHS-2023a OP832526, Meteorus pulchricornis (Wesmael, 1835) NC_053259 (Wei et al. Citation2010), Cotesia flavipes (Cameron, 1891) NC_063945, Microplitis manilae (Ashmead, 1904) MW579537, Cotesia vestalis (Haliday, 1834) NC_014272 (Wei et al. Citation2010), Fopius arisanus (Sonan, 1932) MZ128286 (Cai et al. Citation2022), Psyttalia incisi (Silvestri, 1913) OK413636 (Yang et al. Citation2022), Sigalphus bicolor (Nees, 1812) KF385878, Zele chlorophthalmus (Vollenhoven, 1861) NC_039181 (Wang et al. Citation2020).
Discussion and conclusion
In this study, we successfully sequenced and annotated the complete mitochondrial genome of C. ninghais, making it the first species in the Cyanopterus genus to be sequenced. Phylogenetic analysis indicates that C. ninghais belongs to the subfamily Braconinae within the family Braconidae. Cyanopterus ninghais is closely related to other species in the Braconinae subfamily, including E. yokahamae, V. endoxylaphagus, and H. hebetor, supported by robust Bootstrap values and consistent with morphological classification (Li et al. Citation2021). These findings will provide valuable genetic resources for future research and applications of C. ninghais, and will contribute to a deeper exploration of its ecological significance.
Author contributions
Conception and design: Shaobo Wang, Ke Wei, and Xiaoyi Wang. Sample collection and the molecular experiments: Shaobo Wang and Ke Wei. Drafting of the paper and revising it critically for intellectual content: Shaobo Wang, Ke Wei, and Xiaoyi Wang. All authors reviewed and approved the final manuscript.
Ethical approval
This study does not involve ethical issues. The species used in this study are not endangered organisms, and the sampling site is not located in any protected area, so it did not need any specific permissions. The collection of the insects was legal and reasonable. Information of the voucher specimen and who identified it were included in the manuscript.
Figure S1.jpg
Download JPEG Image (424.4 KB){kind=link}
Table S1.docx
Download MS Word (19.4 KB)README_for_Supplemental material.docx
Download MS Word (15 KB)Figure S2.jpg
Download JPEG Image (419 KB){kind=link}
Acknowledgments
We sincerely thank Dr. Yanlong Tang from Zunyi Normal University for help with the parasitoid colony collection.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov/ under Accession No. OR525636. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA1065096, SRR27556342, and SAMN39450383, respectively.
Additional information
Funding
References
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319. doi:10.1016/j.ympev.2012.08.023.
- Cai PM, Yang DQ, Hao XX, Yue GQ, Jiang LL, Xiao K, Jia PF, Yang JQ, Ji QE, Lin J. 2022. The complete mitochondrial genome of Fopius arisanus (Sonan 1932) (Hymenoptera: Braconidae). Mitochondrial DNA B Resour. 7(1):66–67. doi:10.1080/23802359.2021.2009383.
- Carter JK, Innes P, Goebl AM, Johnson B, Gebert M, Attia Z, Gabani Z, Li R, Melie T, Dart C, et al. 2020. Complete mitochondrial genomes provide current refined phylogenomic hypotheses for relationships among ten Hirundo species. Mitochondrial DNA B Resour. 5(3):2881–2885. doi:10.1080/23802359.2020.1790999.
- Feng ZB, Wu YF, Yang C, Gu XH, Wilson JJ, Li H, Cai WZ, Yang HL, Song F. 2020. Evolution of tRNA gene rearrangement in the mitochondrial genome of ichneumonoid wasps (Hymenoptera: Ichneumonoidea). Int J Biol Macromol. 164:540–547. doi:10.1016/j.ijbiomac.2020.07.149.
- Grant JR, Stothard P. 2008. The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 36(Web Server issue):W181–W184. doi:10.1093/nar/gkn179.
- Huang YX, Qi LQ, Zhang YZ, Jin XX, Wang X. 2020. Sequencing and analysis of the complete mitochondrial genome of Habrobracon hebetor (Hymenoptera: Braconidae). Mitochondrial DNA B Resour. 5(1):1009–1010. doi:10.1080/23802359.2020.1721026.
- Jin JJ, Yu WB, Yang JB, Song Y, DePamphilis CW, Yi TS, Li DZ. 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):241. doi:10.1186/s13059-020-02154-5.
- Kalyaanamoorthy S, Minh BQ, Wong TK, Von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589. doi:10.1038/nmeth.4285.
- Kim W, Byun HW, Jeon MJ. 2022. The complete mitochondrial genome and phylogenetic analysis of Euurobracon yokahamae (Hymenoptera: Braconidae). Mitochondrial DNA B Resour. 7(6):992–993. doi:10.1080/23802359.2022.2080605.
- Li Y, van Achterberg C, Chen XX. 2021. A new genus and eight newly recorded genera of Braconinae Nees (Hymenoptera, Braconidae) from China, with descriptions of fourteen new species. Zookeys. 1038:105–178. doi:10.3897/zookeys.1038.55258.
- Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274. doi:10.1093/molbev/msu300.
- Ronquist F, Teslenko M, Van Der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61(3):539–542. doi:10.1093/sysbio/sys029.
- Wang D, Wang Y, Yu L, Han H, Xu L, Cui Y, Kang A, Pang H. 2020. Sequencing and analysis of the complete mitochondrial genome of Zele chlorophthalmus (Hymenoptera: Braconidae). Acta Entomologica Sinica. 63(8):1028–1038.
- Wang YP, Chen XX, Wu H, He JH. 2009. A new parasitoid (Hymenoptera: Braconidae) of Monochamus alternatus (Coleoptera: Cerambycidae) in China. Biologia. 64(5):942–946. doi:10.2478/s11756-009-0166-8.
- Wei SJ, Shi M, Sharkey MJ, van Achterberg C, Chen XX. 2010. Comparative mitogenomics of Braconidae (Insecta: Hymenoptera) and the phylogenetic utility of mitochondrial genomes with special reference to Holometabolous insects. BMC Genomics. 11(1):371. doi:10.1186/1471-2164-11-371.
- Xu QW, Zhang XJ, Li JX, Ren JR, Ren LL, Luo YQ. 2023a. Pine wilt disease in Northeast and Northwest China: a comprehensive risk review. Forests. 14(2):174. doi:10.3390/f14020174.
- Xu SW, Li WW, Liu QN, Wang YM, Li XL, Duan XQ, He J, Song F. 2023b. The mitochondrial genome of Binodoxys acalephae (Hymenoptera: Braconidae) with unique gene rearrangement and phylogenetic implications. Mol Biol Rep. 50(3):2641–2649. doi:10.1007/s11033-022-08232-0.
- Yang DQ, Hao XX, Jiang LL, Chou TL, Lin X, Yue GQ, Xiao K, Lin J, Ji Q, Cai PM. 2022. The complete mitochondrial genome of Psyttalia incisi (Silvestri, 1916) (Hymenoptera: Braconidae). Mitochondrial DNA B Resour. 7(6):1038–1040. doi:10.1080/23802359.2022.2081942.
- Yuan R-Z, Zhou J-J, Shu X-H, Ye X-Q, Tang P, Chen X-X. 2022. The mitochondrial genome of Chelonus formosanus (Hymenoptera: Braconidae) with novel gene orders and phylogenetic implications. Arch Insect Biochem Physiol. 111(1):e21870. doi:10.1002/arch.21870.
- Zhang D, Gao FL, Jakovlić I, Zou H, Zhang J, Li WX, Wang GT. 2020a. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour. 20(1):348–355. doi:10.1111/1755-0998.13096.
- Zhang X, Li CQ, Pan ZQ, Zhu JC, Wang ZZ, Shi M, Chen XX, Huang JH. 2020b. The complete mitochondrial genome of Asobara japonica (Hymenoptera: Braconidae). Mitochondrial DNA Part B. 5(2):1279–1281. doi:10.1080/23802359.2020.1732238.