
ABSTRACT
Disease-Modifying Anti-Rheumatic Drug (DMARD) use in the treatment of juvenile idiopathic arthritis (JIA) has experienced a dramatic evolution since the early introduction of methotrexate in the 1970’s. This renaissance has been primarily driven by innovation in drug discovery and development that has resulted in the approval of a number of protein-based therapeutics targeting disease-specific pathways. Despite the expansion in the number of drugs available in the treatment of JIA, interindividual variation in therapeutic response and drug-associated toxicities continue to be a major concern and has driven efforts towards individualized therapy. Recent advances in pediatric drug development and innovative approaches to identifying a priori predictors of drug response hold the promise for an individualized approach to therapy that will yield the highest efficacy and safety potential for each JIA patient.
Introduction
Juvenile idiopathic arthritis (JIA) is among the most common chronic diseases of childhood and can result in permanent joint damage with long-term disabilities lasting into adulthood [Citation1]. JIA is estimated to affect approximately 300,000 children in the US and is defined by the development of persistent joint inflammation lasting longer than 6 weeks in patients <16 years of age with no known alternative cause. Further classification of JIA into subtypes is based on several factors, including: number of joints involved, type of joints involved, presence of systemic signs and symptoms, serological markers and family history () [Citation2]. As a result, JIA subtypes are clinically heterogeneous and considered by some to actually represent a myriad of different diseases in childhood presenting with similar symptoms. Despite differences in disease subtype, the general approach to therapy remains similar with the exception of systemic JIA (sJIA), which historically has been resistant to standard DMARD therapy but has more recently been found to be exceptionally responsive to therapeutic agents targeting the pro-inflammatory cytokines IL-1 and IL-6 [Citation3]. This finding is more in line with the realization that this JIA subtype has more clinical similarities to auto-inflammatory diseases also treated with similar therapeutic modalities.
Figure 1. Classification of juvenile idiopathic arthritis by subtype. This schematic illustration highlights the major clinical and laboratory characteristics that are used to differentiate the various subtypes of JIA [Citation2]. The disease subtype can have a significant influence on the patient’s prognosis and treatment approach.
![Figure 1. Classification of juvenile idiopathic arthritis by subtype. This schematic illustration highlights the major clinical and laboratory characteristics that are used to differentiate the various subtypes of JIA [Citation2]. The disease subtype can have a significant influence on the patient’s prognosis and treatment approach.](/cms/asset/81076ee6-fef0-4a7c-bfd2-775ede14e614/tepm_a_1133234_f0001_b.gif)
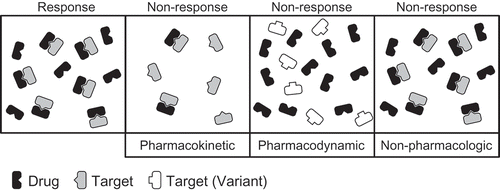
Rapid and aggressive disease control through the early initiation of Disease Modifying Anti-Rheumatic Drug (DMARD) therapy remains the standard of care for the treatment of JIA, in particular polyarticular JIA (pJIA), as duration of untreated disease remains an important contributor to poor long-term outcomes. Despite these efforts, the efficacy of DMARD-based therapeutic regimens remains highly variable and unpredictable, often necessitating modification or escalation of therapy in order to maximize therapeutic outcomes [Citation4]. The delay in response commonly observed with DMARDs represents an additional barrier to achieving early disease control, further complicated by the inability to predict drug outcomes at the onset of treatment. Therefore, major efforts are underway to (i) understand the basis for the observed interindividual variation in response to these drugs and (ii) identify clinical predictors and laboratory biomarkers that can be used to guide clinicians in their choice of drug and dosing. These efforts have included a diversity of approaches and hold the promise of identifying biomarkers to guide clinicians in their efforts towards tailored therapeutic regimens that not only maximize clinical response in patients with a severe disease course, but also minimize overly aggressive treatment in patients with a mild disease course. Such efforts also hold the promise of identifying biomarkers to guide clinicians in predicting risk of relapse upon treatment withdrawal, a process that continues to rely on a trial-and-error approach. Factors associated with the observed variability in response have included specific patient and disease clinical characteristics (e.g. JIA subtypes, disease severity, disease duration) that drive response failure (i.e. non-pharmacologic), as well as differences in how the drug interacts with the body (). This includes both how the drug is handled by the body (i.e. pharmacokinetics) as well as the impact of the drug on the body (i.e. pharmacodynamics). More recent efforts have also taken untargeted and unbiased approaches using omics-based technologies to define biomarkers associated with clinical response.
Figure 2. Contributors to interindividual variability in drug response. This highly simplistic illustration highlights the three major factors believed to drive interindividual variability in drug response. In the response category, sufficient drug is present and it adequately engages its target resulting in clinical response to the drug. In the pharmacokinetic non-response category, an insufficient quantity of drug is present to completely engage its target resulting in sub-optimal response. In the pharmacodynamic non-response category, sufficient drug is present but it is unable to adequately engage its target because of a variation in the target, resulting in failure to respond. Finally, in the non-pharmacologic non-response category, sufficient drug is present and the target is adequately engaged, yet an individual fails to respond to the therapy. This variability is most likely reflective of the heterogeneity of the disease, with different biochemical pathways driving the disease process.
Over the last couple of decades, the biotechnology revolution in the pharmaceutical industry and advancements in pediatric drug development have resulted in the introduction of a plethora of protein-based therapeutics into clinical practice, commonly referred to as biologic DMARDs. These agents primarily target cytokines that have been implicated in the pathogenesis of JIA including: TNFα, IL-1 and IL-6. In contrast to traditional non-biologic DMARDs, these agents have held the promise of targeted therapy and have become increasingly used in the treatment of JIA. Despite the ever increasing use of these agents, the non-biologic DMARD methotrexate (MTX) continues to be the first choice DMARD in the treatment of most forms of JIA. As a result, this work will primarily focus on interindividual variability in response to MTX and many of the more recently introduced biologic DMARDs.
This review will explore the current understanding of factors responsible for the observed interindividual variation in clinical response to DMARDs in the treatment of JIA. The majority of works cited and reviewed in this manuscript were identified through PubMed using a variety of search terms to identify clinical studies that assessed the use of DMARDs in the treatment of JIA and focused primarily on research published within the last 5 years. Initial discussion will focus on barriers to individualized medicine in the pediatric patient population, with a special emphasis on how drugs can behave differently in children as compared with adults and how this can impact drug dosing and response. A discussion of non-biologic and biologic DMARDs will highlight the major differences in these two widely divergent drug classes and how these differences impact their pharmacokinetic and pharmacodynamic properties. Clinical, pharmacokinetic and pharmacodynamic factors associated with variability in clinical response to DMARD therapy will be discussed with regard to how an understanding of these variables can drive efforts towards an individualized approach to drug therapy. Finally, commentary will be provided on more recently adopted omics-based approaches towards the identification of clinical biomarkers that, in the future, may propel the dream of individualized therapy into realization.
Barriers to individualized medicine in pediatrics
Children need to be recognized
Until the 1970’s JIA was primarily seen as an extension of rheumatoid arthritis (RA). However, in the mid 1970’s the Council on Pediatric Rheumatology was formed along with the Pediatric Rheumatology Collaborative Study Group [Citation5], both instrumental in furthering the characterization and treatment of rheumatic diseases in children. It was this collaborative effort that provided the basis for future studies focused on evaluating the safety and efficacy of therapeutic agents in pediatric patients with rheumatic disease. Despite these efforts, children in general continued to represent a vulnerable and marginalized patient population that was often neglected in the drug development process. As a result of extensive advocacy on behalf of all children, congress passed the FDA Modernization Act (FDAMA) of 1997 that incentivized industry-sponsored pediatric drug development by providing an additional 6 months of market exclusivity for conducting such studies and in 1998 the FDA began mandating assessment of new drugs and select marketed drugs in pediatric patients. These provisions were reauthorized in the 2002 Best Pharmaceuticals for Children Act (BPCA) and the 2003 Pediatric Research Equity Act (PREA). Both PREA and BPCA were continued in 2007 with the FDA Amendment Act (FDAAA) and codified in 2012 with the FDA Safety and Innovation Act (FDASIA). The net value of this carrot and stick approach by the US Congress and the FDA to stimulate pediatric drug development can be measured by the dramatic increase in industry-sponsored studies that have led to the FDA indication of numerous new drugs for use in the pediatric population, including many for the treatment of JIA.
The role of ontogeny
The importance of conducting pharmacokinetic and clinical efficacy/toxicity studies in children is evidenced by historical accounts of differences in the disposition and activity of drugs in pediatric patients [Citation6], perhaps most notorious is the cardiovascular collapse in neonates treated with chloramphenicol attributed to the immaturity of major drug clearance pathways in the neonate [Citation7]. This scientific area of investigation, termed developmental pharmacology, evaluates the role of human growth and development on drug disposition and activity. By far, the majority of data collected on the role of ontogeny in drug response focuses on changes in drug disposition, specifically the impact of ontogeny on drug absorption, distribution, metabolism and excretion [Citation8]. These observations have driven the need for comparative pharmacokinetic studies in children during the drug development process in order to target exposure profiles that are similar to those observed in adult studies. The central tenet to these efforts is that the exposure–response relationship does not differ in children as compared with adults. However, in a heterogeneous disease like JIA that shares both similarities and disparities with RA, such assumptions may not hold true, therefore, exposure levels associated with therapeutic response in adults may not necessarily be indicative of the level of drug exposure necessary to elicit similar levels of response in children. This same logic can be applied to any clinical biomarker developed in the adult disease population and argues that data generated in adult disease populations may not be directly translatable to pediatric populations, but rather require further confirmatory investigation.
Children remain ‘biomarker orphans’
Although great strides have been made in pediatric drug development that has greatly expanded our understanding of how drugs behave differently in children, many barriers continue to exist in the area of biomarker development and validation in the pediatric population. These barriers to biomarker development, which form the basis for individualized medicine, has resulted in the coining of the phrase ‘biomarker orphans’ to describe the current state of scientific investigation in pediatric biomarker research [Citation9]. Contributors to this phenomenon include (i) the low prevalence of rheumatic diseases in children forcing multicenter collaborations to obtain a sample size that can offer meaningful results, necessitating studies that are costly and difficult to perform, (ii) the limited pediatric subspecialty workforce whose efforts are often spent providing clinical care to these children rather than spearheading research investigations and (iii) inherent ethical challenges in providing placebo to clinically ill children and/or recruiting healthy controls. These major barriers to research in children are no different than that previously experienced in pediatric drug development, yet need to be overcome to provide safe and effective therapies to children.
DMARDs: non-biologic versus biologic
Traditional, or non-biologic DMARDs, are represented by low-molecular weight drugs from a diversity of pharmacologic classes. Many of the drugs found in this group were initially developed for the treatment of other diseases, but through clinical investigation were found to be effective in the treatment of JIA and other autoimmune diseases. The cornerstone of therapy in the treatment of JIA continues to be MTX, which was originally developed for the treatment of leukemia but later found use in the treatment of autoimmune disease, including JIA [Citation10]. Additional non-biologic DMARDs commonly used in the treatment of JIA and other pediatric rheumatic diseases not covered in this review, include: sulfasalazine, hydroxychloroquine, azathioprine and leflunomide.
The recognition that pro-inflammatory cytokines play a critical role in the pathogenesis of autoimmune arthritis has resulted in the development of protein-based therapeutics targeting these extracellular signaling pathways. Most of these agents were initially developed toward the treatment of RA and other diseases of autoimmune or autoinflammatory etiology. However, many of these agents have been found to be safe and effective in the treatment of JIA. Major targets of this class of drugs that will be the focus of this review will include: TNF-α, IL-1 and IL-6. Despite the fact that all of these agents can be characterized as protein-therapeutics targeted against pro-inflammatory pathways, these agents actually represent a diverse group of drugs including recombinant endogenous proteins, fusion proteins, and monoclonal antibodies, with varying structural, pharmacokinetic and pharmacodynamic properties [Citation11]. These properties impact how these drugs are used in the treatment of JIA and may contribute to the observed variability in response and toxicity.
The most obvious difference between biologic DMARDs and their low-molecular weight counterparts is their mass. For example, the molecular mass of methotrexate is 454.4 Da whereas that of that anti-TNF-α agent adalimumab is ~148,000 Da (). This is greater than twice the mass of human serum albumin and results in dramatic differences in how the drug is delivered to patients, predominantly by the subcutaneous or intravenous route, as well as how the drug distributes and is metabolized within the body [Citation11]. This also presents the precarious issue of antigenic recognition of these foreign proteins resulting in the formation of anti-drug antibodies (ADAs) that can have a dramatic effect on drug response. In addition, as these large molecular weight therapeutics are limited primarily to the vascular space and only enter cells through fluid-phase or receptor mediated endocytosis, they aren’t substrates for the common drug metabolizing enzymes encountered by many of the low-molecular weight therapeutics and avoid many of the common drug–drug and drug–gene interactions encountered with low-molecular weight drugs.
Figure 3. Comparison of biologic and non-biologic DMARDs. The chemical, physical, pharmacokinetic, and pharmacodynamic properties of the biologic DMARD adalimumab and the non-biologic DMARD methotrexate are presented [Citation12,Citation13].
![Figure 3. Comparison of biologic and non-biologic DMARDs. The chemical, physical, pharmacokinetic, and pharmacodynamic properties of the biologic DMARD adalimumab and the non-biologic DMARD methotrexate are presented [Citation12,Citation13].](/cms/asset/1f1d8799-a947-4050-9ee6-12f6c15488d9/tepm_a_1133234_f0003_b.gif)
Effect of clinical factors on response
Early and aggressive therapy
Studies that have evaluated clinical factors associated with improved response to drug therapy have consistently found that early initiation of DMARD therapy is associated with improved therapeutic response, and have further strengthened efforts towards implementation of early and aggressive drug therapy. In a retrospective analysis of 128 JIA patients started on MTX therapy it was found that response to MTX at 6-months was associated with a shorter duration between the diagnosis of JIA and the start of MTX [Citation14]. This finding was supported by a post hoc analysis of the Pediatric Rheumatology International Trials Organization (PRINTO) MTX trial that also found shorter duration of disease prior to initiating MTX therapy as a positive predictor of 6-month response [Citation15]. Studies of drug regimens that include biologic DMARDs targeting TNF-α have similarly demonstrated the importance of early initiation of aggressive therapy in promoting drug response. In a study of early aggressive therapy in JIA (TREAT), an aggressive therapeutic regimen including etanercept (ETN), MTX and a rapid steroid taper was compared with MTX monotherapy in 85 patients with new onset JIA [Citation16]. Although a trend towards attainment of clinically inactive disease was observed in the more aggressive ETN-based regimen, the difference was not statistically significant. However, key findings in this study were that, without regard to the regimen, shorter disease duration prior to treatment and attainment of optimal response by 4 months were associated with an increased duration of clinically inactive disease. In the extension study that followed 48 patients from the initial study for up 2 years, it was found that optimal response at 4 months continued to show a significant association with duration of clinically inactive disease over the study period [Citation17,Citation18]. Findings from an analysis of patients in a German registry that were treated with ETN also support an association between decreased duration of disease prior to initiation of therapy and therapeutic response at 6-months [Citation19]. Together these findings suggest that early initiation of DMARD therapy, independent of drug choice and early control of disease are important predictors of longer term disease control. Determining which drug to use in which patient to achieve early optimal disease control has become the paramount question and future studies will be necessary to establish the risk-benefit ratio of current therapeutic options, also taking into account the higher costs associated with biologic DMARDs.
Disease typing
As compared with non-systemic forms of JIA, which are often responsive to MTX and anti-TNFα therapies, sJIA has been historically resistant to these therapies and current evidence supports a distinct auto-inflammatory etiology that is responsive to drugs targeting IL-1 and IL-6, over MTX and TNF-α [Citation3,Citation20]. This includes early studies that found blood and synovial fluid concentrations of IL-6, but not TNF-α, markedly elevated in patients with sJIA compared with non-systemic JIA and RA [Citation21,Citation22]. However, the association of disease activity with IL-1 levels has been a bit more clouded [Citation23]. Although IL-1 levels were found to be expressed at similar levels in sJIA as healthy individuals, serum from sJIA patients was found to activate peripheral blood mononuclear cells from healthy individuals resulting in pronounced secretion of IL-1β. In addition, it was found that treatment with the anti-IL-1 agent anakinra (i.e. recombinant human IL-1Ra) was found to induce disease remission in 7 of 9 sJIA patients [Citation24]. As a result of these and other studies the most recent American College of Rheumatology treatment recommendations for sJIA emphasizes the earlier use of anti-IL-1 therapies [Citation25–Citation28]. In a retrospective analysis of 262 Dutch JIA patients treated with ETN it was found that patients with sJIA were threefold more likely to experience a poor response to ETN after 15-months of therapy as compared with patients in the non-systemic JIA category [Citation29]. Subsequent investigation of biologic choice following failure of ETN in sJIA showed that the anti-IL1 agent anakinra was superior to other anti-TNF-α therapies, namely infliximab (INF) and adalimumab [Citation30]. As a first line therapy in sJIA, anakinra has also demonstrated increased drug survival and increased induction of inactive disease as compared to ETN [Citation31]. Similarly, the anti-IL-6 agent, tocilizumab (TOC) has been found to be effective in sJIA patients failing initial therapy and has demonstrated efficacy as a first-line agent in the treatment of sJIA despite increased risks of developing severe adverse effects including infection, cytopenias and liver enzyme abnormalities [Citation31–Citation35].
Role of pharmacokinetic variation in drug response
Based on the assumption that interindividual variation in drug response is primarily driven by differences in drug exposure, and the fact that extensive individual pharmacokinetic studies are impractical in clinical practice, the most intuitive and common historical approach towards individualized medicine has been drug level monitoring. These strategies are common clinical practice with a number of narrow therapeutic index drugs, but require great care with regard to the timing of collection respective to dose administration, sample collection and handling, analytical methodologies, and established target levels. Because the clinical value of any biomarker is judged by its association with therapeutic outcomes, it is important that any biomarker, including drug level monitoring, be extensively evaluated in the target population to demonstrate its relationship with drug response and insure its utility in guiding drug treatment decisions.
Erythrocyte MTX polyglutamate levels
Despite a large interindividual variability in the plasma pharmacokinetics of MTX, efforts to associate serum drug levels of MTX with therapeutic response or toxicity have proven unsuccessful [Citation36]. This failure most likely reflects the variable and transient presence of MTX in the serum following once weekly dosing of this short half-life drug [Citation12]. With a reported half-life of approximately 2–4 h, >95% of the drug is cleared within 20 h of dosing. However, the improved understanding in the intracellular metabolism of MTX into pharmacologically active polyglutamated metabolites resulted in assessment of these metabolites in circulating erythrocytes and peripheral blood mononuclear cells with evidence that increased levels of these metabolites were associated with drug response in RA [Citation37]. Follow-up studies on the association of erythrocyte levels of MTX polyglutamates (MTXGlun) with clinical response in RA have been inconsistent, but these assays remain available to clinicians in the care of their patients [Citation38–Citation41]. A cross-sectional study of erythrocyte MTXGlun levels in the JIA patient population has found these levels to display a high level of variability with a >45-fold variation in measured levels (range: 4.8–218.5 nmol/l) [Citation42]. Increased accumulation of long-chain polyglutamates (i.e. 3–5 glutamate residues) was associated with increased dose and the subcutaneous route of administration, but not with reduced disease activity. However, increased long-chain polyglutmate levels were found to be associated with an increased incidence of gastrointestinal side effects and elevated liver enzymes. An earlier cross-sectional study of erythrocyte MTXGlun in JIA patients actually found that MTXGlun levels were higher in patients categorized as non-responders compared with responders, but the difference was not significant [Citation43]. More recently, a prospective analysis of erythrocyte MTXGlun after 3-months of MTX therapy found that increased MTXGlun levels were associated with reduced disease activity scores at both 3- and 12-months [Citation44]. These findings are encouraging that erythrocyte MTXGlun levels are associated with improved clinical response, however further studies are needed to establish drug level targets that differentiate optimal and sub-optimal exposure. Additionally, once these targets are set, additional analysis of their benefit as a tool in therapeutic drug monitoring will need to be established through prospective evaluation before they are ready for routine clinical practice.
Immunogenicity and therapeutic drug monitoring with biologic DMARDs
The landmark study by Maini et al. of INF in the treatment of RA demonstrated several key features of biologic DMARD therapy that are believed to be generally applicable to this class of drugs and are the focus of many ongoing efforts [Citation45]. First, this study demonstrated that INF given in combination with MTX is synergistic and results in greater efficacy than either drug given alone. Second, it demonstrated that the development of ADAs is associated with reduced therapeutic response and appears to be inhibited by concomitant MTX therapy. Lastly, it demonstrated that lower doses of INF are associated with an increase in ADA formation. In agreement with these findings, efficacy studies of INF in JIA found that doses of 3 mg/kg as compared with 6 mg/kg were associated with a threefold increased risk of developing ADAs, a reduction in serum trough INF levels to undetectable levels in >50% of the patients by 44 weeks of therapy and a twofold increase in the incidence of serious adverse events [Citation46]. Increased serum trough INF levels were found to be associated with clinical response and were increased in patients not developing ADAs. Similarly, formation of ADAs to the anti-TNF-α drug adalimumab were observed in 26% of a small cohort of JIA patients (n = 23) over 52 months and the presence of ADAs were found to be associated with an 8.7-fold reduction in trough adalimumab levels compared with the ADA-negative patients [Citation47]. Patients receiving MTX were significantly less likely to develop ADAs and 83% of ADA-positive patients developed loss of response compared with 6% of ADA-negative patients. Genetic susceptibility to risk of ADA formation has been suggested in recent studies that found that an Ashkenazi Jewish ancestry was protective of ADA formation, with a relative risk reduction on par with concomitant DMARD therapy [Citation48]. Together these studies highlight the potential importance of therapeutic drug level and ADA monitoring in patients receiving anti-TNF-α therapy and is evidenced by their routine monitoring in many patient care settings. As all therapeutic proteins possess the risk for the development of immunogenicity, these findings suggest that in the future many of these same principles may be applied to other therapeutic proteins. The consistent finding that MTX and other non-biologic DMARDs play a critical role in reducing ADA formation is an important area of ongoing study that has recently been reviewed elsewhere [Citation49]. The more broad utilization of MTX in non-rheumatic disease states to preserve response to therapeutic proteins may be an extreme notion, but such efforts may be born out as MTX is increasingly utilized in other disease states like inflammatory bowel disease (IBD), where INF is heavily used and loss of therapeutic response to INF is a major concern.
Age-dependent clearance of biologic DMARDs
Pharmacokinetic analysis of INF in JIA patients has demonstrated an inverse relationship between age and plasma clearance [Citation50]. The finding that INF clearance rates were directly related to resting energy expenditure in this pediatric cohort suggests that younger children and patients with increased metabolic rates may experience more rapid clearance of INF, therefore requiring either an increase in INF dose or a shorter interval between dosing. Pharmacokinetic studies on tocilizumab (TOC) have also found increased drug clearance in children that is reflected in the weight-based dosing found in its labeling. In a recent study of TOC in subjects with pJIA, pre-dose serum trough levels were measured at 16 weeks following every 4 week dosing. Subjects weighing ≥30 kg receiving 8 mg/kg TOC had, on average, serum trough levels 7.6-fold higher than patients <30 kg receiving the same 8 mg/kg dosing, and 2.7-fold higher than patients <30 kg receiving 10 mg/kg [Citation51]. In support of age-dependent changes in clearance of these macromolecular-weight drugs, increased therapeutic protein clearance in young children has also been documented for anakinra and the IL-1β monoclonal antibody canakinumab [Citation52,Citation53]. The role of interindividual variation in drug exposure resulting from differences in age-dependent drug clearance rates remains an area in need of further research, as these factors drive age- and weight-dependent dosing in drug efficacy studies and can have a significant impact on observed therapeutic response.
Role of pharmacodynamic variation in response
Although MTX was not originally developed for the treatment of autoimmune disease, it does represent an early example of a rationally designed drug developed to inhibit folate-dependent biochemical pathways in the treatment of human disease [Citation54]. Despite the wealth of data on the pharmacological properties of MTX, its mechanism of action in the treatment of JIA and other autoimmune diseases remains poorly understood and controversial [Citation55]. Although an understanding of how MTX elicits its pharmacologic activity in JIA remains poorly defined, an in-depth understanding of the biochemical pathways targeted by MTX allows for investigation for the role of differential inhibition of these pathways on the observed variability in response. Similarly, biologic DMARDs were developed to target specific dysregulated immune and inflammatory pathways that are amenable to investigation of their role in the observed variability in drug response.
Folates in MTX therapy
Since MTX is a highly potent inhibitor of dihydrofolate reductase, perhaps the most obvious pharmacodynamic measure of MTX activity would be changes in circulating folate concentrations. The potential of measuring circulating folate concentrations as a measure of MTX activity is supported by a recent cross-sectional study of folate levels in JIA patients using a liquid chromatography-tandem mass spectrometry analytical assay that measured the major circulating bioactive folate species, 5-methyl-tetrahydrofolate (5mTHF) in erythrocytes [Citation56]. Median 5mTHF levels in patients receiving MTX for at least 3 months were more than 30% lower than those measured in individuals not receiving MTX. Among patients receiving MTX, 5mTHF levels displayed a high degree of interindividual variability with plasma levels ranging 11.5-fold. Interestingly, folic acid supplementation was documented in 47% of patients receiving MTX, but was not found to be associated with any difference in circulating 5mTHF levels. In contrast to these findings, two recent studies in RA patients that utilized competitive folate binding protein immunoassays to measure serum and erythrocyte folate found that initiation of MTX therapy along with folic acid supplementation at levels of 10–30 mg/week resulted in no significant reduction in serum or erythrocyte folate levels [Citation57,Citation58]. The origin of this discrepancy may reflect differences in the level of folate supplementation in the studies, innate differences in systemic folate regulation in pediatric versus adult patients, or more than likely, differences in the analytical methodologies marked by the inability of competitive folate binding protein assays to distinguish between individual folate species with a higher affinity of these proteins for the inactive supplemental form of folate (i.e. folic acid) as compared with the major circulating biologically active form (i.e. 5mTHF) [Citation59]. However, it is interesting to note that associations between higher baseline erythrocyte folate levels and lower disease activity scores after 3 months of MTX therapy was observed in one of the studies [Citation57] and reduced serum folates levels at baseline were associated with ‘undesirable symptoms’ related to MTX therapy in the other [Citation58]. These findings require further study to determine the potential role of monitoring circulating folate levels in the treatment of JIA, in particular, studies are needed to see if variation in folate levels can account for the observed variations in drug efficacy and toxicity. Additional downstream markers of inhibition of folate-dependent pathways, including metabolites in the purine and pyrimidine biosynthesis pathways warrant further study to evaluate their potential association with MTX response and toxicity [Citation60].
Pro-inflammatory protein markers of response
Application of multiplex cytokine analyses to JIA plasma and synovial fluid samples has demonstrated distinct pro-inflammatory protein expression profiles that differ between active and inactive disease, as well as appear to show distinct signatures of systemic-onset, oligoarticular and polyarticular JIA [Citation21,Citation61–Citation64]. Further studies are needed to understand how cytokine profiles can be used to stratify patient populations in the early optimization of drug therapy, especially with the increased utilization of agents targeting specific cytokine pathways. In contrast to JIA patients treated with MTX alone, those receiving combination MTX+INF experienced a significant and sustained reduction in circulating IL-6, ICAM-1 and MMP-3 by 2 weeks of therapy [Citation65]. However, clinical response was only associated with reductions in ICAM-1 and MMP-3. Recent investigations have targeted plasma IL-1β, IL-6, and TNF-α levels as therapeutic markers in JIA patients receiving ETN [Citation64]. Only IL-6 levels were found to decrease following the initiation of ETN and to be positively correlated with disease activity scores. Although previously reported, it is interesting to note that initiation of ETN was associated with a dose-dependent increase in median TNF-α levels of ~200-fold, however, this expanded pool of TNF-α appears to be bound to ETN and inactive, and has been supported in other studies of ETN and INF [Citation62,Citation66]. A similar effect has been observed with acute elevations in serum IL-6 and sIL-6 R levels following initiation of TOC and has been attributed to binding of TOC to sIL-6 R and the IL-6 receptor resulting in decreased receptor-mediated IL-6 clearance and increased circulating sIL-6 R-TOC complexes [Citation32,Citation67,Citation68]. These findings limit the clinical utility of measuring serum cytokine levels in patients receiving biologic DMARDs as a measure of targeted anti-cytokine activity, but may represent the opportunity to assess assays that differentiate free sIL-6 R and TNF-α from their drug complexes as a pharmacodynamic indicator.
Although increased disease activity has been consistently associated with elevated plasma IL-6 concentrations, anti-IL-6 therapy has been mostly reserved for the treatment of Sjia [Citation69]. However, a recent study in pJIA patients with inadequate response to MTX, TOC was found to be effective with clinical response recorded within 16 weeks of therapy in 89% of patients [Citation51]. This response was further evaluated in a placebo-controlled 24-week withdrawal phase that found a reduced incidence of flare in patients receiving TOC compared with placebo (25.6 vs 48.1%, p = 0.002). These findings highlight the possible role of identifying dysregulated pathways in JIA in the selection of currently available drugs towards individualization of therapy, but also show the importance of understanding the disease pathophysiology in identifying druggable targets.
Myeloid-related protein 8 (MRP8) and MRP14 are calcium-binding proteins secreted by activated phagocytic cells that form a heterocomplex (MRP8/14), also referred to as calprotectin. MRP8/14 is a pro-inflammatory mediator that binds and activates Toll-like receptor-4 and its levels in serum have been associated with disease activity in JIA patients [Citation70,Citation71]. In children discontinued on MTX therapy 6 or 12 months after disease remission, elevated MRP8/14 levels at the time of discontinuation were significantly associated with an increased risk of disease relapse [Citation72]. The resulting area under the receiver operator curve for prediction of 3-month relapse was 0.76, with a sensitivity of 87% and a specificity of 64%. A similar prognostic value has been demonstrated in a cohort of sJIA patients, where levels were significantly decreased with response, and elevated levels predicted flares with a sensitivity of 92% and specificity of 88% [Citation73]. The prognostic value of serum MRP8/14 levels has been demonstrated for both MTX and anti-TNF-α therapies [Citation74,Citation75]. Consistent with increased therapeutic response to MTX in patients with higher disease activity, patients with elevated baseline MRP8/14 levels were found to be more likely to respond to MTX [Citation74]. Similarly, in patients treated with anti-TNF-α therapy, elevated MRP8/14 levels at baseline were predictive of clinical response, while sustained elevations in MRP8/14 at treatment discontinuation were associated with increased risk of relapse [Citation75]. Such findings support the role of serum MRP8/14 monitoring as a prognostic indicator of drug response and risk of relapse.
Distinctions between anti-TNF-α therapies
In recent data on the long-term safety of anti-TNF-α therapy that included ETN without MTX, ETN with MTX, MTX alone and adalimumab in JIA it was found that ETN without MTX was associated with an increased incidence of uveitis and IBD [Citation76]. These findings are in line with several studies that support an increased incidence of IBD and uveitis in JIA patients treated with ETN [Citation77–Citation79]. The relationship between ETN and the incidence of IBD and uveitis requires further investigation, but it is interesting to note that adalimumab and INF, which have the same molecular target, are recommended in the treatment of uveitis in JIA [Citation80] and indicated in the treatment of pediatric IBD. Pharmacodynamic differences among the anti-TNF-α therapies are also supported by some level of clinical response to adalimumab or INF following failure of ETN [Citation30,Citation81,Citation82]. It has been suggested that this variation in clinical response may reflect differences in the binding characteristics of these agents [Citation83]. Binding studies of INF and ETN to soluble TNF-α (sTNF-α) and transmembrane TNF-α (tTNF-α) found that INF binds monomeric sTNF-α (inactive), trimeric sTNF-α (active), and tTNF-α; while ETN binds the monomeric sTNF-α and tTNF-α [Citation84]. In addition, it was found that INF complexation with sTNF-α was much more stable than that observed with ETN and that TNF-α dissociating from ETN was biologically active [Citation84]. Additionally, differences in the ability of adalimumab and INF to induce complement-dependent cytotoxicity (CDC) has been suggested as a source for the variation in pharmacologic response, but studies in activated PBMCs have failed to show CDC with any of these agents [Citation85]. As a result, it is clear that all anti-TNF-α therapies are not the same and that additional studies are needed to understand what is the basis for the observed differences in clinical response to these agents despite a common therapeutic target.
Omics approach to biomarker identification
With the increased availability of complementary and overlapping omics-based technologies it is becoming important to utilize these orthogonal methods to identify and validate the role of specific genetic, epigenetic and metabolic pathways in drug response. Specifically, the use of untargeted approaches coupled with the ability to perform network analyses on these large datasets will provide valuable information as we try to unravel the mechanistic basis for observed variations in drug response. Although these tools have more recently found use in the investigation of personalized approaches to medicine, it is anticipated that the use and integration of these technologies will only expand.
Genetic biomarkers
Pharmacogenomic approaches towards the determination of the role of genetic variation on MTX response in JIA has recently been reviewed elsewhere [Citation86]. The majority of these efforts have used targeted pharmacogenetic strategies to evaluate the relationship between polymorphisms of candidate genes and clinical response to MTX. These studies have demonstrated relationships between variation in genes involved in the disposition of MTX, including members of the ATP-binding cassette (ABC) superfamily of transporters ABCB1, ABCC3, ABCC1, as well as the reduced folate carrier (SLC19A1) and the proton-coupled folate transporter(SLC46A1) [Citation87–Citation89]. Similarly, evaluations of polymorphisms of genes involved in biochemical pathways targeted by MTX have yielded associations with clinical response and include enzymes involved in regulation of cellular folate and purine nucleotide pools such as methylentetrahydrofolate reductase (MTHFR), methionine synthase reductase (MTRR), inosine triphosphatepyrophosphatase (ITPA) and aminoimidazolecarboxamide ribonucleotide formyltransferase (ATIC) [Citation90,Citation91]. A MTX non-response prediction model was developed and validated using two JIA cohorts and included polymorphisms in ABCB1, ABCC1, SLC46A1 and MTRR [Citation87]. With the increasing availability of genotyping in clinical practice, the potential for genotypic prediction of clinical outcomes is enormous, but will require additional independent validation before such findings can be routinely implemented.
Non-targeted whole genome analysis was conducted in 694 JIA patients to identify genetic polymorphisms associated with response to MTX and identified a number of genes [Citation92]. The strongest association was observed with a gene that encodes the alpha chain of a low-voltage calcium channel important in neuronal signaling (CACNA1I). Additional loci identified included the cystic fibrosis transmembrane conductance regulator (CFTR) gene, and two genes related to TGFβ signaling (ZMIZ1 and TGIF1). Although these polymorphisms do not directly replicate previous targeted genetic studies, it is interesting to note that CFTR, also known as ABCC7, is a member of the ABC superfamily of transporters that have previously been implicated as an important factor in MTX transport and response. The finding that genes involved in TGFβ signaling also supports previous studies of the role of this pathway as important in MTX response [Citation93]. The difficulty with these untargeted studies is the risk of false positives given the large number of variables measured in a relatively small population, as well as the inability to rationalize the basis and practicality for the observed associations. A benefit of this approach is that it allows for the identification of biochemical pathways potentially involved in drug response that would not have otherwise been considered. However, orthogonal approaches are needed to validate the role of these pathways in drug response. Regardless, any such findings require further study to replicate and validate their relevance and such studies may be viewed more as hypothesis-generating, rather than hypothesis-driven.
Transcriptomics
Measurement of isolated leukocyte gene expression profiles has found that transcript levels for a number of genes are differentially regulated in sJIA as compared to healthy controls and patients with non-sJIA systemic inflammatory conditions [Citation94]. This study focused primarily on the use of this platform as a diagnostic tool but found that initiation of anakinra in eight patients resulted in a change in expression of over 389 transcripts. Although the transcripts affected by anakinra only partially overlapped with those identified as diagnostic transcripts, these findings present the possibility that gene expression profiling could be utilized as a tool to monitor response to therapy in these patients. Subsequent study of blood gene expression profiling in a cohort of sJIA patients treated with anakinra found significant changes in 522 transcripts following 1 month of therapy [Citation95]. A module-level analysis of grouped genes was used to follow response to anakinra and found changes in transcript modules associated with platelets, cytotoxicity, erythropoiesis, and IFN-inducible genes. A study of differential gene expression in a group of 11 JIA patients following the initiation of MTX was able to demonstrate a significant change in over 1222 gene transcripts [Citation93]. By narrowing the analysis to only patients achieving maximal response to MTX, the transcript pool was further narrowed to 6 genes of interest for which subsequent genotyping in 123 JIA patients identified three polymorphisms in SLC16A7 that were associated with non-response to MTX. Although these findings require further investigation to validate the relationship of this gene with MTX response and the mechanistic basis of any such interaction, the strength of this study is that it utilized gene expression profiling to identify genes for subsequent targeted genotyping analysis. A more recent study evaluated whole blood gene expression profiles in pJIA patients and healthy control subjects [Citation96]. Differential expression of 90 genes between healthy controls and pJIA patients were observed and included a number of genes previously associated with immunologic, inflammatory, and connective tissue disease. Further, support vector machines were used to build predictive models of disease activity at 6- and 12-months using gene transcript levels at baseline. These findings support a relationship between baseline gene expression profiles and likelihood of clinical response, suggesting that such an approach may be amenable to patient stratification and prediction of probability of drug response, towards the goal of individualized medicine.
Proteomics
Proteomics is another promising technology that has yet to be used to identify predictive clinical biomarkers of response in JIA. However, recent data in 22 RA patients treated with ETN and MTX used a targeted mass spectrometry-based proteomic approach to assess MRP8 (S100A8) and MRP14 (S100A9) levels in patient PBMC and serum samples [Citation97]. Baseline MRP14 levels were significantly elevated in both PBMCs and serum samples of patients classified as responders at 6 months as compared to non-responders, whereas for MRP8, only PBMC levels were elevated in the response group. In a follow-up study an untargeted proteomics approach was used to identify baseline protein biomarkers of response in RA patients treated with ETN and MTX [Citation98]. With the identification of 213 proteins in the initial cohort of 22 patients, 12 proteins were found to be differentially expressed in patients classified as responders at 6 months. These findings were further validated in an independent cohort of 16 RA patients and demonstrated that increased expression of a number of acute phase proteins at baseline are predictive of response, most notably complement component 7 and vitamin K-dependent protein S.
Metabolomics
Although this approach also has not been applied to studies in the JIA population, it holds great promise as a tool in identifying clinical biomarkers of drug response. A study using nuclear magnetic resonance (NMR) spectroscopy-based metabolic profiling found that the metabolomic profile of patients with RA differ from healthy individuals, and that the profile in patients with active RA differ from those in remission, suggesting the possible role of metabolomic profiling in clinical monitoring [Citation99]. Identified metabolites include lactate and acetylated glycoproteins, which are associated with inflammation and joint damage, as well as an increase in circulating cholesterol that is associated with decreased HDL levels that is characteristic of RA. A mass spectrometry-based approach towards identifying metabolomic-driven diagnostics for RA have also found significant differences in RA patients as compared to healthy subjects that were notable for changes in nucleotide, cholesterol, and amino acid metabolism [Citation100]. A more recent NMR-based study of RA patients and healthy subjects has continued to support the role of metabolomics as a discriminatory tool in the diagnosis and monitoring of therapy in RA [Citation101,Citation102]. Metabolomic profiling was able to differentiate RA patients from healthy controls, as well as RA patients with active disease from those in remission. NMR-based metabolomic analysis of serum samples obtained at baseline in newly diagnosed RA patients treated with MTX monotherapy were able to identify 11 major metabolites that discriminated responders from non-responders after 24 weeks of drug therapy [Citation103]. As compared to non-responders, responders had significantly elevated levels of uric acid, taurine, histidine, methionine, glycine, hypoxanthine, and significantly lower levels of uracil, trimethylamine-N-oxide, α-oxoglutarate, aspartate, and trypthophan. These findings suggest that differences in response to MTX are associated with variations in several metabolic pathways, including nucleotide metabolism, one-carbon metabolism and the citric acid cycle.
Expert commentary
With the recent expansion of DMARDs available for the treatment of JIA, and several currently in development, clinicians may find themselves overwhelmed with options without sufficient evidence-based data to guide them in drug selection and dosing. It is clear that some patients appear to respond to some therapies better than others and the basis for these variations in response remain mostly unknown and often result in delayed disease control and prolonged exposure to ineffective and potentially toxic drugs. Therefore, it is imperative to understand the factors responsible for the observed variation in response to DMARDs in the treatment of JIA so that patients can be stratified based on validated clinical and laboratory measures early in the disease process to insure treatment with drug regimens that have the highest probability of inducing disease remission. Great strides have been made in stratifying patients based on disease subtype that have been found to have a profound effect on patient response to therapy. However, additional efforts are needed to understand how variation in the disposition and pharmacological activity of drugs can impact clinical response and yield measureable clinical biomarkers that can be used to guide the drug selection and dosing decision process.
Five-year view
In the next five years work will focus on the discovery of novel biomarkers of drug response utilizing untargeted and unbiased omics-based approaches. These efforts will allow the integration of multiple layers of genomic, transcriptomic, and metabolomic data. Despite the untargeted approach, this hypothesis generating and pragmatic approach to biomarker discovery will embrace advanced statistical and biochemical network analysis tools to build an understanding of how drugs interact with the body and how these interactions influence the disease process and can be used in the stratification and routine care of our patients. These efforts are expected to address the questions identified in this review related to the variability in physiologic pathways governing DMARD exposure in JIA, as well as variation in the biochemical pathways targeted by these individual drugs. Hypotheses generated through these approaches will be tested and validated in both pre-clinical models and prospective patient cohorts, as are currently being conducted for the preliminarily identified biomarkers discussed in this review. This integration of pre-clinical and clinical approaches is mandatory to substantiate the biochemical validity of observed associations and to verify the findings in clinical practice settings. Research questions addressed using this approach will also focus on understanding the biochemical pathways that differentiate JIA disease subtypes in the identification of druggable targets, and differentiate responders and non-responders to specific drug regimens towards the development of therapeutic stratification approaches in the treatment of JIA.
In addition to ongoing efforts to establish current biomarkers focused primarily on drug efficacy, an increased focus on biomarkers of drug-related toxicities is expected. Drug-associated toxicities are often predictable based on the known pharmacologic properties of the drug and are likely to be reflected in interindividual variability in pharmacokinetic and pharmacodynamic measures. Lastly, in JIA, a large degree of interindividual variation in drug efficacy most likely reflects the heterogeneity of the disease and therefore may not be associated with measurable pharmacokinetic or pharmacodynamic differences. Ongoing characterization of the clinical features of JIA and validation of clinical outcome measures continues to be a priority of the pediatric rheumatology community on a global level. Multinational collaborative efforts strive to move the field forward and will continue to advance the field in the years ahead.
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Key Issues
Clinical response to DMARD therapy in the treatment of JIA is variable, delayed and unpredictable which makes it an ideal candidate for individualized medicine.
Development of clinical biomarkers towards the implementation of individualized medicine in the pediatric population is restricted by many of the same issues that have historically faced pediatric drug development, most notably a small patient population, and will require a collaborative and concerted effort to move the field forward.
Non-biologic and biologic DMARDs represent two distinct classes of drugs with unique physicochemical and pharmacological properties. Through an understanding of how these differences influence drug disposition and activity, potential factors affecting drug response can be identified.
Clinical descriptive factors, most notably disease subtype classification, are important contributors to the observed variability in drug response.
Variability in the disposition of non-biologic and biologic DMARDs can account for some of the observed variability in response, and has resulted in the implementation of therapeutic drug monitoring for anti-TNF-α therapies and some evidence for monitoring of erythrocyte MTXGlun levels.
An understanding of the pathophysiology of disease can help identify biomarkers for monitoring disease response to therapy, as well as assist in drug selection.
Assessment of laboratory markers of biochemical response to drug therapy as an indicator and predictor of drug response is an ongoing effort, but has yielded promising results particularly with MRP8/14.
Expansion of omics-based approaches to biomarker identification in the treatment of JIA is expected to yield a deeper understanding of variables affecting drug response and to provide clinically meaningful and measureable laboratory markers to guide drug therapy.
References
- Helmick CG, Felson DT, Lawrence RC, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Arthritis Rheum. 2008;58:15–25.
- Petty RE, Southwood TR, Manners P, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. 2004;31:390–392.
- Correll CK, Binstadt BA. Advances in the pathogenesis and treatment of systemic juvenile idiopathic arthritis. Pediatr Res. 2014;75:176–183.
- Becker ML. Optimization of pediatric rheumatology therapeutics. Clin Pharmacol Ther. 2012;91:597–606.
- Schaller JG. The history of pediatric rheumatology. Pediatr Res. 2005;58:997–1007.
- Kearns GL, Abdel-Rahman SM, Alander SW, et al. Developmental pharmacology–drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349:1157–1167.
- Weiss CF, Glazko AJ, Weston JK. Chloramphenicol in the newborn infant. A physiologic explanation of its toxicity when given in excessive doses. N Engl J Med. 1960;262:787–794.
- Funk RS, Brown JT, Abdel-Rahman SM. Pediatric pharmacokinetics: human development and drug disposition. Pediatric Clinics of North America. 2012;59:1001–1016.
- Kearns GL. Beyond biomarkers: an opportunity to address the ‘pharmacodynamic gap’ in pediatric drug development. Biomark Med. 2010;4:783–786.
- Giannini EH, Brewer EJ, Kuzmina N, et al. Methotrexate in resistant juvenile rheumatoid arthritis. Results of the U.S.A.-U.S.S.R. double-blind, placebo-controlled trial. The Pediatric Rheumatology Collaborative Study Group and The Cooperative Children’s Study Group. N Engl J Med. 1992;326:1043–1049.
- Xu Z, Davis HM, Zhou H. Rational development and utilization of antibody-based therapeutic proteins in pediatrics. Pharmacol Ther. 2013;137:225–247.
- Balis FM, Savitch JL, Bleyer WA. Pharmacokinetics of oral methotrexate in children. Cancer Research. 1983;43:2342–2345.
- Humira [package insert]. North Chicago, IL: AbbVie Inc.; 2015.
- Albers HM, Wessels JA, van der Straaten RJ, et al. Time to treatment as an important factor for the response to methotrexate in juvenile idiopathic arthritis. Arthritis Rheum. 2009;61:46–51.
- Vilca I, Munitis PG, Pistorio A, et al. Predictors of poor response to methotrexate in polyarticular-course juvenile idiopathic arthritis: analysis of the PRINTO methotrexate trial. Ann Rheum Dis. 2010;69:1479–1483.
- Wallace CA, Giannini EH, Spalding SJ, et al. Trial of early aggressive therapy in polyarticular juvenile idiopathic arthritis. Arthritis Rheum. 2012;64:2012–2021.
- Wallace CA, Ringold S, Bohnsack J, et al. Extension study of participants from the trial of early aggressive therapy in juvenile idiopathic arthritis. J Rheumatol. 2014;41:2459–2465.
- Wallace CA, Giannini EH, Spalding SJ, et al. Clinically inactive disease in a cohort of children with new-onset polyarticular juvenile idiopathic arthritis treated with early aggressive therapy: time to achievement, total duration, and predictors. J Rheumatol. 2014;41:1163–1170.
- Geikowski T, Becker I, Horneff G, et al. Predictors of response to etanercept in polyarticular-course juvenile idiopathic arthritis. Rheumatology. 2014;53:1245–1249.
- Woo P, Southwood TR, Prieur AM, et al. Randomized, placebo-controlled, crossover trial of low-dose oral methotrexate in children with extended oligoarticular or systemic arthritis. Arthritis Rheum. 2000;43:1849–1857.
- De Benedetti F, Massa M, Robbioni P, et al. Correlation of serum interleukin-6 levels with joint involvement and thrombocytosis in systemic juvenile rheumatoid arthritis. Arthritis Rheum. 1991;34:1158–1163.
- De Benedetti F, Pignatti P, Gerloni V, et al. Differences in synovial fluid cytokine levels between juvenile and adult rheumatoid arthritis. J Rheumatol. 1997;24:1403–1409.
- De Benedetti F, Pignatti P, Massa M, et al. Circulating levels of interleukin 1 beta and of interleukin 1 receptor antagonist in systemic juvenile chronic arthritis. Clin Exp Rheumatol. 1995;13:779–784.
- Pascual V, Allantaz F, Arce E, et al. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005;201:1479–1486.
- Nigrovic PA, Mannion M, Prince FH, et al. Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: report of forty-six patients from an international multicenter series. Arthritis Rheum. 2011;63:545–555.
- Hedrich CM, Bruck N, Fiebig B, et al. Anakinra: a safe and effective first-line treatment in systemic onset juvenile idiopathic arthritis (SoJIA). Rheumatol Int. 2012;32:3525–3530.
- Ringold S, Weiss PF, Beukelman T, et al. update of the 2011 American College of Rheumatology recommendations for the treatment of juvenile idiopathic arthritis: recommendations for the medical therapy of children with systemic juvenile idiopathic arthritis and tuberculosis screening among children receiving biologic medications. Arthritis Rheum. 2013;65:2499–2512.
- Vastert SJ, De Jager W, Noordman BJ, et al. Effectiveness of first-line treatment with recombinant interleukin-1 receptor antagonist in steroid-naive patients with new-onset systemic juvenile idiopathic arthritis: results of a prospective cohort study. Arthritis Rheumatol. 2014;66(4):1034–1043.
- Otten MH, Prince FH, Armbrust W, et al. Factors associated with treatment response to etanercept in juvenile idiopathic arthritis. JAMA. 2011;306:2340–2347.
- Otten MH, Prince FH, Anink J, et al. Effectiveness and safety of a second and third biological agent after failing etanercept in juvenile idiopathic arthritis: results from the Dutch National ABC Register. Ann Rheum Dis. 2013;72:721–727.
- Woerner A, Uettwiller F, Melki I, et al. Biological treatment in systemic juvenile idiopathic arthritis: achievement of inactive disease or clinical remission on a first, second or third biological agent. RMD Open. 2015;1(1):e000036.
- Woo P, Wilkinson N, Prieur AM, et al. Open label phase II trial of single, ascending doses of MRA in Caucasian children with severe systemic juvenile idiopathic arthritis: proof of principle of the efficacy of IL-6 receptor blockade in this type of arthritis and demonstration of prolonged clinical improvement. Arthritis Res Ther. 2005;7:R1281–1288.
- Yokota S, Miyamae T, Imagawa T, et al. Therapeutic efficacy of humanized recombinant anti-interleukin-6 receptor antibody in children with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2005;52:818–825.
- Yokota S, Imagawa T, Mori M, et al. Efficacy and safety of tocilizumab in patients with systemic-onset juvenile idiopathic arthritis: a randomised, double-blind, placebo-controlled, withdrawal phase III trial. Lancet. 2008;371:998–1006.
- De Benedetti F, Brunner HI, Ruperto N, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367:2385–2395.
- Ravelli A, Di Fuccia G, Molinaro M, et al. Plasma levels after oral methotrexate in children with juvenile rheumatoid arthritis. J Rheumatol. 1993;20:1573–1577.
- Angelis-Stoforidis P, Vajda FJ, Christophidis N. Methotrexate polyglutamate levels in circulating erythrocytes and polymorphs correlate with clinical efficacy in rheumatoid arthritis. Clin Exp Rheumatol. 1999;17:313–320.
- Dervieux T, Furst D, Lein DO, et al. Polyglutamation of methotrexate with common polymorphisms in reduced folate carrier, aminoimidazole carboxamide ribonucleotide transformylase, and thymidylate synthase are associated with methotrexate effects in rheumatoid arthritis. Arthritis Rheum. 2004;50:2766–2774.
- Dervieux T, Furst D, Lein DO, et al. Pharmacogenetic and metabolite measurements are associated with clinical status in patients with rheumatoid arthritis treated with methotrexate: results of a multicentred cross sectional observational study. Ann Rheum Dis. 2005;64:1180–1185.
- Stamp LK, O’Donnell JL, Chapman PT, et al. Methotrexate polyglutamate concentrations are not associated with disease control in rheumatoid arthritis patients receiving long-term methotrexate therapy. Arthritis Rheum. 2010;62:359–368.
- Stamp LK, Barclay ML, O’Donnell JL, et al. Effects of changing from oral to subcutaneous methotrexate on red blood cell methotrexate polyglutamate concentrations and disease activity in patients with rheumatoid arthritis. J Rheumatol. 2011;38:2540–2547.
- Becker ML, Gaedigk R, van Haandel L, et al. The effect of genotype on methotrexate polyglutamate variability in juvenile idiopathic arthritis and association with drug response. Arthritis Rheum. 2011;63:276–285.
- Dolezalova P, Krijt J, Chladek J, et al. Adenosine and methotrexate polyglutamate concentrations in patients with juvenile arthritis. Rheumatology. 2005;44:74–79.
- Calasan MB, den Boer E, de Rotte MC, et al. Methotrexate polyglutamates in erythrocytes are associated with lower disease activity in juvenile idiopathic arthritis patients. Ann Rheum Dis. 2015;74:402–407.
- Maini RN, Breedveld FC, Kalden JR, et al. Therapeutic efficacy of multiple intravenous infusions of anti-tumor necrosis factor alpha monoclonal antibody combined with low-dose weekly methotrexate in rheumatoid arthritis. Arthritis Rheum. 1998;41:1552–1563.
- Ruperto N, Lovell DJ, Cuttica R, et al. A randomized, placebo-controlled trial of infliximab plus methotrexate for the treatment of polyarticular-course juvenile rheumatoid arthritis. Arthritis Rheum. 2007;56:3096–3106.
- Skrabl-Baumgartner A, Erwa W, Muntean W, et al. Anti-adalimumab antibodies in juvenile idiopathic arthritis: frequent association with loss of response. Scand J Rheumatol. 2015;44:359–362.
- Ungar B, Haj-Natour O, Kopylov U, et al. Ashkenazi Jewish origin protects against formation of antibodies to infliximab and therapy failure. Medicine (Baltimore). 2015;94:e673.
- Jani M, Barton A, Warren RB, et al. The role of DMARDs in reducing the immunogenicity of TNF inhibitors in chronic inflammatory diseases. Rheumatology. 2014;53:213–222.
- Goldman JL, Davis HM, Zhou H, et al. Infliximab clearance in children: potential association with resting energy expenditure. Ann Paediatr Rheum. 2012;1:120–125.
- Brunner HI, Ruperto N, Zuber Z, et al. Efficacy and safety of tocilizumab in patients with polyarticular-course juvenile idiopathic arthritis: results from a phase 3, randomised, double-blind withdrawal trial. Ann Rheum Dis. 2015;74:1110–1117.
- Ruperto N, Quartier P, Wulffraat N, et al. A phase II, multicenter, open-label study evaluating dosing and preliminary safety and efficacy of canakinumab in systemic juvenile idiopathic arthritis with active systemic features. Arthritis Rheum. 2012;64:557–567.
- Urien S, Bardin C, Bader-Meunier B, et al. Anakinra pharmacokinetics in children and adolescents with systemic-onset juvenile idiopathic arthritis and autoinflammatory syndromes. BMC Pharmacol Toxicol. 2013;14(1):40.
- Ward JR. Historical perspective on the use of methotrexate for the treatment of rheumatoid arthritis. J Rheumatol Suppl. 1985;12(Suppl 12):3–6.
- Chan ES, Cronstein BN. Methotrexate–how does it really work? Nat Rev Rheumatol. 2010;6:175–178.
- Funk RS, Van Haandel L, Leeder JS, et al. Folate depletion and increased glutamation in juvenile idiopathic arthritis patients treated with methotrexate. Arthritis Rheumatol. 2014;66(12):3476–3485.
- De Rotte MC, de Jong PH, Pluijm SM, et al. Association of low baseline levels of erythrocyte folate with treatment nonresponse at three months in rheumatoid arthritis patients receiving methotrexate. Arthritis Rheum. 2013;65:2803–2813.
- Dhir V, Sandhu A, Kaur J, et al. Comparison of two different folic acid doses with methotrexate–a randomized controlled trial (FOLVARI Study). Arthritis Res Ther. 2015;17(1):156.
- Nygren-Babol L, Sternesjo A, Jagerstad M, et al. Affinity and rate constants for interactions of bovine folate-binding protein and folate derivatives determined by optical biosensor technology. Effect of stereoselectivity. J Agric Food Chem. 2005;53:5473–5478.
- Funk RS, Van Haandel L, Becker ML, et al. Low-dose methotrexate results in the selective accumulation of aminoimidazole carboxamide ribotide in an erythroblastoid cell line. J Pharmacol Exp Ther. 2013;347:154–163.
- de Jager W, Hoppenreijs EP, Wulffraat NM, et al. Blood and synovial fluid cytokine signatures in patients with juvenile idiopathic arthritis: a cross-sectional study. Ann Rheum Dis. 2007;66:589–598.
- Spirchez M, Samasca G, Iancu M, et al. Relation of interleukin-6, TNF-alpha and interleukin-1alpha with disease activity and severity in juvenile idiopathic arthritis patients. Clin Lab. 2012;58:253–260.
- Shimizu M, Nakagishi Y, Yachie A. Distinct subsets of patients with systemic juvenile idiopathic arthritis based on their cytokine profiles. Cytokine. 2013;61:345–348.
- Kaminiarczyk-Pyzalka D, Adamczak K, Mikos H, et al. Proinflammatory cytokines in monitoring the course of disease and effectiveness of treatment with etanercept (ETN) of children with oligo- and polyarticular juvenile idiopathic arthritis (JIA). Clin Lab. 2014;60:1481–1490.
- Visvanathan S, Wagner C, Marini JC, et al. The effect of infliximab plus methotrexate on the modulation of inflammatory disease markers in juvenile idiopathic arthritis: analyses from a randomized, placebo-controlled trial. Pediatr Rheumatol Online J. 2010;8:24.
- Charles P, Elliott MJ, Davis D, et al. Regulation of cytokines, cytokine inhibitors, and acute-phase proteins following anti-TNF-alpha therapy in rheumatoid arthritis. J Immunology. 1999;163:1521–1528.
- Uchiyama Y, Yoshida H, Koike N, et al. Anti-IL-6 receptor antibody increases blood IL-6 level via the blockade of IL-6 clearance, but not via the induction of IL-6 production. Int Immunopharmacol. 2008;8:1595–1601.
- Zhang X, Morcos PN, Saito T, et al. Clinical pharmacology of tocilizumab for the treatment of systemic juvenile idiopathic arthritis. Expert Rev Clin Pharmacol. 2013;6:123–137.
- De Benedetti F, Robbioni P, Massa M, et al. Serum interleukin-6 levels and joint involvement in polyarticular and pauciarticular juvenile chronic arthritis. Clin Exp Rheumatol. 1992;10:493–498.
- Frosch M, Strey A, Vogl T, et al. Myeloid-related proteins 8 and 14 are specifically secreted during interaction of phagocytes and activated endothelium and are useful markers for monitoring disease activity in pauciarticular-onset juvenile rheumatoid arthritis. Arthritis Rheum. 2000;43:628–637.
- Frosch M, Ahlmann M, Vogl T, et al. The myeloid-related proteins 8 and 14 complex, a novel ligand of toll-like receptor 4, and interleukin-1beta form a positive feedback mechanism in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2009;60:883–891.
- Foell D, Wulffraat N, Wedderburn LR, et al. Methotrexate withdrawal at 6 vs 12 months in juvenile idiopathic arthritis in remission: a randomized clinical trial. Jama. 2010;303:1266–1273.
- Holzinger D, Frosch M, Kastrup A, et al. The Toll-like receptor 4 agonist MRP8/14 protein complex is a sensitive indicator for disease activity and predicts relapses in systemic-onset juvenile idiopathic arthritis. Ann Rheum Dis. 2012;71:974–980.
- Moncrieffe H, Ursu S, Holzinger D, et al. A subgroup of juvenile idiopathic arthritis patients who respond well to methotrexate are identified by the serum biomarker MRP8/14 protein. Rheumatology. 2013;52:1467–1476.
- Anink J, Van Suijlekom-Smit LWA, Otten MH, et al. MRP8/14 serum levels as a predictor of response to starting and stopping anti-TNF treatment in juvenile idiopathic arthritis. Arthritis Res Ther. 2015;17(1):200.
- Klotsche J, Niewerth M, Haas J-P, et al. Long-term safety of etanercept and adalimumab compared to methotrexate in patients with juvenile idiopathic arthritis (JIA). Ann Rheum Dis. 2015. doi:10.1136/annrheumdis-annrheumdis-2014-206747. [Epub ahead of print].
- Ruemmele FM, Prieur AM, Talbotec C, et al. Development of Crohn disease during anti-TNF-alpha therapy in a child with juvenile idiopathic arthritis. J Pediatr Gastroenterol Nutr. 2004;39:203–206.
- Prince FH, Twilt M, ten Cate R, et al. Long-term follow-up on effectiveness and safety of etanercept in juvenile idiopathic arthritis: the Dutch national register. Ann Rheum Dis. 2009;68:635–641.
- Van Dijken TD, Vastert SJ, Gerloni VM, et al. Development of inflammatory bowel disease in patients with juvenile idiopathic arthritis treated with etanercept. J Rheumatol. 2011;38:1441–1446.
- Levy-Clarke G, Jabs DA, Read RW, et al. Expert panel recommendations for the use of anti-tumor necrosis factor biologic agents in patients with ocular inflammatory disorders. Ophthalmology. 2014;121:785–796 e783.
- Katsicas MM, Russo RA. Use of adalimumab in patients with juvenile idiopathic arthritis refractory to etanercept and/or infliximab. Clin Rheumatol. 2009;28:985–988.
- Schmeling H, Minden K, Foeldvari I, et al. Efficacy and safety of adalimumab as the first and second biologic agent in juvenile idiopathic arthritis: the German Biologics JIA Registry. Arthritis Rheumatol. 2014;66(9):2580–2589.
- Mpofu S, Fatima F, Moots RJ. Anti-TNF-alpha therapies: they are all the same (aren’t they?). Rheumatology. 2005;44:271–273.
- Scallon B, Cai A, Solowski N, et al. Binding and functional comparisons of two types of tumor necrosis factor antagonists. J Pharmacol Exp Ther. 2002;301:418–426.
- Kaymakcalan Z, Sakorafas P, Bose S, et al. Comparisons of affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin Immunol. 2009;131:308–316.
- Pastore S, Stocco G, Favretto D, et al. Genetic determinants for methotrexate response in juvenile idiopathic arthritis. Front Pharmacol. 2015;6:52.
- Bulatovic M, Heijstek MW, Van Dijkhuizen EH, et al. Prediction of clinical non-response to methotrexate treatment in juvenile idiopathic arthritis. Ann Rheum Dis. 2012;71:1484–1489.
- De Rotte MC, Bulatovic M, Heijstek MW, et al. ABCB1 and ABCC3 gene polymorphisms are associated with first-year response to methotrexate in juvenile idiopathic arthritis. J Rheumatol. 2012;39:2032–2040.
- Pastore S, Stocco G, Moressa V, et al. 5-Aminoimidazole-4-carboxamide ribonucleotide-transformylase and inosine-triphosphate-pyrophosphatase genes variants predict remission rate during methotrexate therapy in patients with juvenile idiopathic arthritis. Rheumatol Int. 2015;35:619–627.
- Schmeling H, Biber D, Heins S, et al. Influence of methylenetetrahydrofolate reductase polymorphisms on efficacy and toxicity of methotrexate in patients with juvenile idiopathic arthritis. J Rheumatol. 2005;32:1832–1836.
- Hinks A, Moncrieffe H, Martin P, et al. Association of the 5-aminoimidazole-4-carboxamide ribonucleotide transformylase gene with response to methotrexate in juvenile idiopathic arthritis. Ann Rheum Dis. 2011;70:1395–1400.
- Cobb J, Cule E, Moncrieffe H, et al. Genome-wide data reveal novel genes for methotrexate response in a large cohort of juvenile idiopathic arthritis cases. Pharmacogenomics J. 2014;14:356–364.
- Moncrieffe H, Hinks A, Ursu S, et al. Generation of novel pharmacogenomic candidates in response to methotrexate in juvenile idiopathic arthritis: correlation between gene expression and genotype. Pharmacogenetics and Genomics. 2010;20:665–676.
- Allantaz F, Chaussabel D, Stichweh D, et al. Blood leukocyte microarrays to diagnose systemic onset juvenile idiopathic arthritis and follow the response to IL-1 blockade. J Exp Med. 2007;204:2131–2144.
- Quartier P, Allantaz F, Cimaz R, et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann Rheum Dis. 2011;70:747–754.
- Jiang K, Sawle AD, Frank MB, et al. Whole blood gene expression profiling predicts therapeutic response at six months in patients with polyarticular juvenile idiopathic arthritis. Arthritis Rheumatol. 2014;66(5):1363–1371.
- Obry A, Lequerré T, Hardouin J, et al. Identification of S100A9 as biomarker of responsiveness to the methotrexate/etanercept combination in rheumatoid arthritis using a proteomic approach. Plos One. 2014;9(12):e115800.
- Obry A, Hardouin J, Lequerre T, et al. Identification of 7 proteins in sera of RA patients with potential to predict ETA/MTX treatment response. Theranostics. 2015;5:1214–1224.
- Lauridsen MB, Bliddal H, Christensen R, et al. 1H NMR spectroscopy-based interventional metabolic phenotyping: a cohort study of rheumatoid arthritis patients. J Proteome Res. 2010;9:4545–4553.
- Madsen RK, Lundstedt T, Gabrielsson J, et al. Diagnostic properties of metabolic perturbations in rheumatoid arthritis. Arthritis Res Ther. 2011;13(1):R19.
- Kapoor SR, Filer A, Fitzpatrick MA, et al. Metabolic profiling predicts response to anti-tumor necrosis factor alpha therapy in patients with rheumatoid arthritis. Arthritis Rheum. 2013;65:1448–1456.
- Young SP, Kapoor SR, Viant MR, et al. The impact of inflammation on metabolomic profiles in patients with arthritis. Arthritis Rheum. 2013;65:2015–2023.
- Wang Z, Chen Z, Yang S, et al. (1)H NMR-based metabolomic analysis for identifying serum biomarkers to evaluate methotrexate treatment in patients with early rheumatoid arthritis. Exp Ther Med. 2012;4:165–171.