
ABSTRACT
Cystic fibrosis (CF) affects about 70,000 individuals worldwide, whose lives are shortened mainly due to chronic pulmonary infections resulting from impaired clearance of abnormally viscous airway mucus. The development of novel drugs targeting specific CFTR gene mutations in a precision medicine framework improved treatment, so that for patients born in 2000–2003 in the UK, the median life expectancy was estimated at around 40 years. Moreover the discovery of the CRISPR (Classes of Regularly Interspaced Palindromic Repeats) and Cas9 (Crispr-ASsociated) nuclease system opened the perspective of specifically correcting the defective CFTR gene as recently demonstrated in a model of intestinal stem cell organoids from CF patients. In the present review, we shall outline the existing state-of-art treatments and the perspectives for the precision treatment of CF opened by CRISPR.
Introduction
Cystic fibrosis (CF) is a life-shortening autosomal recessive condition, caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene resident on the long arm of chromosome 7 (7q31.2) that encodes for a chloride ion channel regulated by cAMP-dependent phosphorylation (). This gene comprises 27 exons spanning over 190 kb. After splicing of the large introns, the CFTR mRNA comprises 6.5 kb.[Citation1] All the exons are required to produce a functional protein but different mutations have different impacts, which is why six classes of CFTR mutation have been defined. Clinical severity of CF is determined by the interactions between CFTR mutation class, modifier genes and the environment [Citation2] Recent advances demonstrating that the CFTR gene does not operate in isolation but rather in the dynamic network of interacting components that impact its protein structure, folding, stability etc. led to the emergence of a new concept of CF functional landscape (CFFL).[Citation3] These different factors are connected to a protein fold management system called the proteostasis network. In this respect the sum of the interactions between CFTR and the protein ‘social network’ in the CFFL determine the severity of the disease. Furthermore, given that it is not always easy to quantify the relative contribution of environmental and genetic factors in CF, it may be possible that even correcting CFTR to a certain extent may not be sufficient to completely cure the disease, in cases where environmental factors play a large role.[Citation2] Therefore a deeper understanding of these interactions may lead to a shift of the clinical management of cystic fibrosis toward more personalized approaches. Indeed, clinical improvements become apparent when at least 10% of ‘normal’ CFTR gene activity is reached, which may be achieved in certain situations by various interventions or combinations thereof, opening therefore the perspective for precision medicine diagnostic and treatment strategies to be proposed. A recent phase III study showed that ivacaftor 150 mg twice a day lead to a decrease in sweat chloride of approximately 50 mmol/l, corresponding with a 10–12% improvement in forced expiratory volume in one second (FEV1) [Citation4] but in principle clinical improvements become measurable once a certain threshold level of CFTR activity is achieved by the treatment combinations.[Citation5]
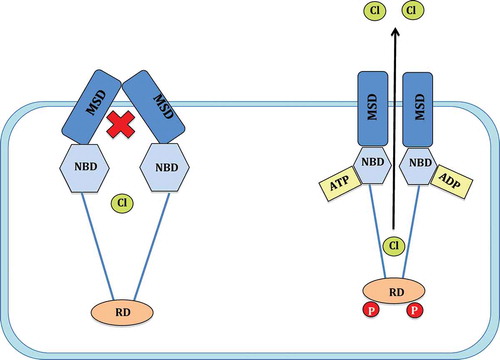
Figure 1. Mechanism of CFTR channel gating. The CFTR channel consists of two membrane spanning domains (MSD), two nucleotide binding domains (NBD) and a single regulatory domain (RD). Following the phosphorylation of the RD by protein kinase A (PKA) and the hydrolysis of an ATP on the NBD, the CFTR channel gate moves to an open conformation and allows the passage of chloride (Cl−) ions (adapted from reference 6, Chen et al, 2006 with kind permission from S. Karger AG, Basel).
The recent discovery of the CRISPR (Classes of Regularly Interspaced Palindromic Repeats) and Cas9 (Crispr-ASsociated) nuclease system opened the perspective of specifically targeting genes for the purpose of correcting them or modulating their expression. Most importantly, it has been recently shown that using the CRISPR/Cas9 system it is possible to repair the dysfunctional dystrophin gene in Duchenne muscular dystrophy patient stem cells [Citation6,Citation7] and the CFTR gene in intestinal stem cell organoids from CF patients.[Citation8] This progress opens the perspective of undertaking the precise correction of the CFTR gene in the frame of a precision medicine approach. This use of organoids represents a step toward the use of these models for precision medicine, as these model structures have been used in the past predominantly for personalized medicine. Indeed personalized therapies are underlined by stratification that results, in some cases, from the use of tests carried out in patient-derived intestinal or pulmonary organoids. These organoids allow the preclinical testing of new drug combinations in order to identify which ones are most effective for the individual patient from whose cells the organoid was generated and in this respect concur to the delivery of personalized medicine.[Citation9] Conversely, precision medicine, whilst having the identical ultimate aim of achieving the best possible treatment for patients as personalized medicine, relies upon the initial identification of the genotypic, environmental and phenotypic components that drive patients disease and their response to treatments, based on large databases and ‘big data’ rather than patient-specific organoid testing.
In this review we shall summarize the current understanding of CF pathophysiology and present the precise classification of CFTR mutation classes. We shall then describe the corresponding mutation class-directed precise treatments for CFTR (e.g. aminoglycosides, ataluren, amlexanox, CFTR potentiators such as ivacaftor, CFTR correctors such as lumacaftor and VX-661 etc.). We shall finish by outlining the potential advantages of using targeted nucleases for treating CF and the obstacles that need overcoming in this respect.
Overview of CF pathophysiology
CFTR is a member of the ATP-binding cassette (ABC) family typically resident on epithelia.[Citation10] Production of a mature, functional CFTR ion channel requires DNA transcription; mRNA translation at the endoplasmic reticulum (ER); post-translational modifications at the Golgi complex and finally translocation to and insertion within the epithelial cell membrane. Mature CFTR consists of five subunits: two membrane-spanning domains (MSD); two nucleotide binding domains (NBD); and a unique regulatory domain (RD). CFTR is a cAMP-dependent ion channel, as RD phosphorylation through cAMP-dependent protein kinase A (PKA) is a necessary prerequisite for subsequent channel gating by ATP. The hydrolysis of a single adenosine triphosphate (ATP) occurs within the NBD ATP-binding site.
The disruption to the CFTR chloride ion channel protein product effectively diminishes CFTR-dependent chloride secretion at all secretory epithelial surfaces. Secondary effects on epithelial sodium (Na) concentrations and subsequent upsurge in epithelial Na channel (ENaC) activity enhance Na reabsorption and promote excessive fluid loss at the epithelial surface. This dehydrated epithelial coating confers significant, adverse effects upon the airways and also the exocrine function, resulting in many of the complications observed in CF patients across multiple organs, spanning respiratory, gut, pancreas and reproductive systems. Furthermore, there is increasing evidence that defective bicarbonate transport represents a major problem in cystic fibrosis.[Citation11]
The pulmonary system is significantly affected in CF, alongside pancreatic and vas deferens dysfunction, with respiratory failure persisting as a leading cause of mortality in sufferers. In the lungs, mucociliary clearance is impaired because of hyperviscosity of the fluid airway surface layer (ASL) and submucosal gland secretions.[Citation12] triggering repeat cascades of inflammation, recurrent infection and bronchiectasis, with irreparable damage resulting in mortality. Infertility is often apparent in males with CF because of a congenital absence of the vas deferens. Exocrine and endocrine pancreatic insufficiency also frequently features, resulting in steatorrhoea with nutritional deficiencies and CF-related diabetes mellitus.[Citation1]
CF sufferers additionally display hypertonic sweat chloride concentrations (above 600 mmol/l) because of the failure of chloride ion transport into sweat glands. Sweat chloride concentrations have been shown to correlate with CFTR activity level [Citation13] and are therefore most commonly utilized within CF diagnosis [Citation1] and as a measure of response to treatment within clinical trials investigating the use of new agents. Recent work by McKone et al. [Citation14] portrays sweat chloride level as being independently associated with survival in CF patients where CFTR genotype is unclassified. However early death from cystic fibrosis in the first decade of life is now rare because of improving clinical management. Considering the perspectives of introducing new therapies, it is very likely that the life expectation of newly diagnosed CF patients may increase in the future.[Citation15] Alternatively, nasal potential difference (NPD) measurements can be obtained for use in diagnosis and the assessment of respiratory epithelium-specific response to CFTR therapeutics.
CFTR mutation classes
To date, nearly 2000 mutations have been identified as causative of CF.[Citation16] Mutations are classed I–VI according to precise effects on the CFTR product [Citation1] and have been linked to disease severity, though all manifest in the fundamental absence of chloride transport at the epithelial surface ().
Figure 2. A schematic representation of class I to class VI mutations in cystic fibrosis. Class I mutations lead to a complete lack of production of the CFTR protein. Class II mutations result in defective handling of CFTR, typically stimulating ER retention and degradation. Class III and class IV mutations disrupt CFTR function by decreasing its gating ability and conductance respectively. Defective splicing of pre-mRNA to mRNA typically underlies class V mutations. Finally, class VI mutations increase the turn-over of CFTR at the cell membrane.
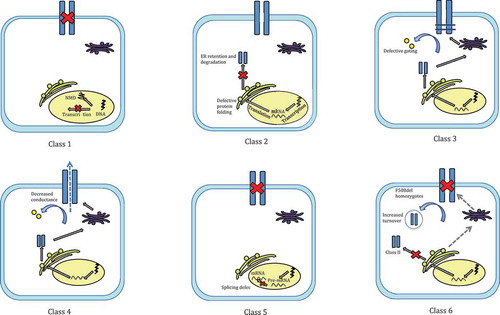
Class I mutations, such as G542X, R553x, W1282X and R1162X [Citation16], result in the near or complete absence of CFTR at the epithelial surface. Class I mutations are most commonly nonsense mutations positioned within the CFTR DNA coding region, causing premature termination codons (PTC) when transcribed to mRNA. PTC constitute the majority of all described CF-causing mutant alleles and are present in 5–8% of CF patients. Frameshift mutations and flaws in mRNA splicing are less common causes.[Citation17] The resultant protein is truncated or deformed and with mRNA frequently subject to nonsense mediated decay (NMD), with those evading NMD being non-functional.
Class II mutations are the result of defective protein processing, resulting in CFTR misfolding and abnormalities in protein maturation.[Citation18] The majority of the dysfunctional protein product is subject to rapid degradation within the ER, however some of this protein does appear to reach the cell surface.[Citation19]
Class III mutations confer functional defects, relating to dysfunctional CFTR gating in response to agonists at the apical membrane. The most common class III mutation, G551D, is a missense mutation caused by alterations to ATP binding pocket structure.[Citation20]
Class IV mutations commonly reside in the DNA sequence which codes for MSDs.[Citation17] CFTR present in the plasma membrane retain ‘gating’ activity and exhibit inefficient chloride conductivity. Class IV mutations therefore more commonly, though not uniformly, represent a milder form of CF with partial function maintained depending on the nature of the conduction defect (as partial or full).
Class V and VI mutations are relatively less severe, respectively the result of defective pre-mRNA splicing and reduced CFTR protein lifetime at the epithelial membrane. While class V and VI mutations generally reduce CFTR concentration within the plasma membrane through distinct means, with class V mutations impairing correct RNA splicing mechanisms and class VI mutations affecting CFTR quality and subsequently, stability. Unstable CFTR reaching the cell surface is subject to accelerated turnover and removal.
F508del mutation
F508del results from phenylalanine deletion at position 508, and is the most prevalent CFTR mutation, possessed by over 90% of CF patients worldwide. F508del is considered a class II.[Citation21] III and VI [Citation22] mutation. Class IV–VI mutations are typically milder than the severe F508del homozygous genotype, because of the relative maintenance of chloride passage. Wide variation in CF lung disease severity across the F508del homozygous population illustrates the additional contribution of environmental factors and regulatory genes to the CF phenotype.
Mutation class-directed precise treatments for CFTR
Presently, various pharmacological agents are available to enhance the expression of a functional CFTR protein. CFTR modulators are specific molecules targeted toward select CFTR defects, promoting full expression and functionality. Modulators include read-through agents, correctors and potentiators.
Treatments that promote the read-through of premature termination codons
Read-through agents overcome aberrant stop codons, resulting in the production of a full-length, functional CFTR protein.
Aminoglycosides
Aminoglycosides act upon binding to the 18S ribosomal subunit, subverting normal ribosomal DNA proofreading activity to permit the incorporation of an alternative amino acid at the post-transcriptional control (PTC) level. The aminoglycoside G418 increases the expression of the cAMP-dependent CFTR in HeLa cells with G542X or R553X nonsense mutations.[Citation23] The level of CFTR on HeLa cell surfaces correlated to the dose of G418 administered and was up to 35% of that observed in a wild-type control.[Citation24]
Cell treatment with gentamycin resulted in the display of full-length CFTR. Gentamycin had a greater effect in patients who were homozygous for nonsense mutations, which is likely as a result of gentamycin inducing the read-through of two mutated CFTR alleles, leading to greater protein expression.
Gentamycin enhanced the read-through of the relatively rare Y122X mutation in vitro, and this correlated with in vivo findings of increased chloride transport through CFTR, reductions in sweat chloride levels and improved respiratory function, as measured by FEV1 and FVC, evidencing a clinical benefit of aminoglycosides. IV administration of gentamycin has provided additional support for its use in CF patients with nonsense mutations.[Citation25] In keeping with previous findings, there were no detectable differences in the patients who were homozygous for the F508del mutation.
However, not all data support aminoglycoside use for CF patients with nonsense mutations. Following nasal administration of gentamycin and tobramycin, Clancy et al. found no significant improvement in ion transport across the nasal epithelium and no increase of plasma membrane CFTR.[Citation26] The studies in which gentamycin was reported to enhance CFTR read-through used higher doses and aimed to achieve peak serum concentrations of 20–40 µg/ml, as opposed to 8–10 µg/ml in studies reporting no effect. This suggests gentamycin’s efficacy in PTC suppression is dose-dependent [Citation23,Citation25] and highlights the importance of the route of administration.
Ataluren
The non-aminoglycoside ataluren (PTC124) has up to 15 times more potency than other read-through agents [Citation27] and was shown to improve lung function in patients not taking inhaled aminoglycosides. This made ataluren an ideal treatment for patients with nonsense mutations. A phase II clinical trial including 19 CF patients with at least one nonsense mutation, found modest increases in total chloride transport and CFTR function over the 12 week study period.[Citation28] A non-statistically significant increase in pulmonary function was noted, possibly because of the small sample size. Subsequent phase III clinical trials [Citation29] found that the change in FEV1 from baseline at the end of the 48 week trial was not significantly different between the two groups (−2.5% ataluren compared with −5.5% placebo, p = 0.12) and the rate of pulmonary exacerbations were similar between groups. The change in the FEV1 from baseline at the end of the 48 week trial was not significantly different between the two groups (−2.5% ataluren compared with −5.5% placebo, p = 0.12) and the rate of pulmonary exacerbations were similar between groups. Interestingly, 15% of CF patients who received ataluren possessed elevated creatinine concentrations, in comparison with <1% of the placebo group, thus contesting the safety of ataluren. Regardless of the controversy surrounding its safety, Ataluren was shown to significantly improve lung function in those patients who were not taking inhaled aminoglycosides.
Amlexanox
The anti-inflammatory agent amlexanox has dual capabilities as NMD inhibitor and inducer of PTC read-through.[Citation30] Amlexanox increased both the number of mRNA transcripts containing stop codons and full-length CFTR protein in HeLa cells expressing a nonsense mutation. Future research should establish its efficacy and compare this to ataluren and aminoglycoside treatment.
CFTR potentiators
The small molecules termed ‘CFTR potentiators’ modulate CFTR function at the plasma membrane, increasing the open probability in response to agonists. They are therefore the ideal therapeutic candidates for the 5% of CF patients possessing class III ‘gating’ mutations, with additional interest apparent in the domain of class II mutations, such as F508del. Furthermore, potentiators may act to improve chloride conductance in class IV mutations, whereby ion flow could be increased through activation of additional channels at the cell surface.
Ivacaftor
Ivacaftor (also known as VX-770 or Kalydeco) is the most clinically successful CFTR potentiator to date, acting primarily on class III gating mutations. Initial identification was through high-throughput screening of over 200,000 compounds.[Citation31] In vitro patch-clamp studies of both mutant Fisher rat thyroid (FRT) and G551D/F508del heterozygotic HBE mutant cells demonstrated that ivacaftor addition increased chloride ion conductance from ~5 to 50%. This was correlated to an increase in the channel open probability of the CFTR channel. Addition of amiloride, an ENaC inhibitor, had no effect on results, demonstrating that observed outcomes were not as a consequence of ENaC modulation. Ivacaftor did not amplify chloride conductance to the same extent in F508del homozygotes, as they display a reduced quantity of CFTR channels capable of potentiation present at the cell surface.
The STRIVE and ENVISION 48 week phase III placebo-controlled trials further established the clinically beneficial role of ivacaftor in patients with the G551D mutation. STRIVE investigated the response to ivacaftor in 161 individuals aged 12 and over, with an FEV1 > 40% predicted. Results were encouraging, with FEV1 improving by a mean value of 5% in three-fourth of adults enrolled, also linked to sustained reductions in sweat chloride levels.[Citation32] Further positive observations included a reduction in the number of pulmonary exacerbations, an increase in weight and a patient-verified improvement. ENVISION had a similar study design as STRIVE but investigated 6–11 years old children.[Citation33] Comparable changes in FEV1, pulmonary exacerbations, weight gain and patient-recorded outcomes were reported.
A recent randomized clinical trial conducted by Moss et al. [Citation34] investigated the effect of 24 weeks of ivacaftor treatment in patients with the Arg117His CFTR mutation. Non-significant improvements in percent-predicted FEV1 were reported, however, when adults were specifically analyzed, significant improvements in their predicted FEV1 were demonstrated. In addition to this, sweat chloride levels were reduced in all groups and this was accompanied by a general improvement in self-reported overall health and well-being. Recent reports from the open-label study, PERSIST, which followed on from the STRIVE and ENVISION trials, assessed the safety profile of 150 mg of ivacaftor bi-daily for 96 weeks. Continued improvements in FEV1, weight and pulmonary exacerbations were noted throughout the study period and no new safety concerns arose.[Citation35]
Information regarding the precise mechanism of action of ivacaftor and other potentiators is lacking. It is thought that CFTR potentiators do not act to increase regulatory domain (RD) phosphorylation as ivacaftor was seen to potentiate CFTR channel opening independently of RD status.[Citation35] Additional research by Yeh et al. [Citation36] and Bompadre et al. [Citation37] indicates that ivacaftor works on areas other than the second NBD. Mutations of the ATP binding pocket on NBD1, however, were seen to have dramatic effect on the potentiating ability of the potentiator P-ATP, suggesting that this is its binding site.[Citation38] The observation that ivacaftor is equally efficacious at potentiating the CFTR channel regardless of which side of the membrane it is presented, supports the hypothesis that ivacaftor may work at more than one site of action.[Citation36]
Moreover, the Food and Drug Administration (FDA) has approved ivacaftor for use in CF patients aged six and above with G551D and 10 other CFTR gating mutations. Initial suggestions that ivacaftor can act to enhance ion transport in vitro in F508del homozygous mutations have not been duplicated in vivo.[Citation38] Ivacaftor treatment in these patients slightly improved sweat chloride levels, but did not significantly improve FEV1. The enhanced chloride transport suggests that the small amount of F508del at the plasma membrane is responsive to ivacaftor, as seen in previous in vitro studies.[Citation31] This suggests that ivacaftor treatment may be successful if a CFTR corrector is firstly applied to prevent CFTR misfolding, thus enhancing the number of molecules available for ivacaftor to potentiate. Precision medicine approaches would be optimal for defining the precise mechanism of action of ivacaftor and other potentiators, as this may identify individual CFTR mutations which would respond greatest to individual potentiators.
CFTR correctors
CFTR correctors enhance the delivery of CFTR to the cell surface in F508del and other class II mutations by promoting ER passage. They appear to act by two distinct mechanisms. Compounds which bind directly to the CFTR protein, encouraging its appropriate folding and maturation, are termed ‘pharmacological chaperones’. Alternatively, the corrector may modulate any of the multiple phases of protein regulation and are thus labeled ‘proteostasis regulators’. Therefore ‘proteostasis regulators’ may influence additional protein synthesis pathways, in addition to CFTR.
Corrector molecules were initially identified following the screening of over 150,000 chemical compounds.[Citation39] Measurements of iodide and chloride flux were coupled with observations of a fluorescent halide molecule in FRT cells expressing F508del CFTR. Successful correctors increased the amount of mature, glycosylated CFTR and in turn enhanced iodide and chloride flux through the channel. Pulse-chase analysis confirmed that correctors did not alter transcription or translation of CFTR and possible potentiator actions of the molecules were eliminated. Hence, the corrector compounds identified facilitated the correct processing and maturation of CFTR, allowing its display on the cell surface. The most efficacious corrector, as identified in HBE cells, Corr-4a, was able to increase chloride secretion through CFTR by up to 8% of that measured in wild-type.
Lumacaftor
A potent CFTR corrector lumacaftor, also known as VX-809, has recently been discovered.[Citation40] Results suggest that lumacaftor acts to increase the folding and stability of the CFTR protein by directly binding to it, acting as a CFTR chaperone.[Citation40] Application of lumacaftor to cells expressing F508del resulted in a sixfold increase in the release of CFTR from the ER, suggesting that CFTR conformation is more resistant to degradation. Furthermore, the use of brefeldin A, an inhibitor of protein trafficking from the ER, implied that lumacaftor creates a CFTR form which is resistant to ER degradation, as the rate of decay of CFTR in the ER was slower in lumacaftor treated cells in comparison with controls. This transpired not to be as a result of inhibition of protein breakdown, but rather subsequent to enhanced stability of CFTR. Finally, the action of CFTR at the cell surface appeared normal.
All these findings indicate that lumacaftor enhances the correct folding and promotes the stability of CFTR, with it then resisting ER breakdown and progressing to incorporation into the plasma membrane. A significant limitation to the above study, is that lumacaftor did not correct CFTR to normal levels, the reason for this remaining undiscovered. Notwithstanding this limitation, recent work by Ren et al. [Citation41] has supported the hypothesis that lumacaftor works as a CFTR chaperone, with findings that lumacaftor improves interactions between the NBD and MSD to improve folding. Interestingly, the correcting effect of lumacaftor was boosted by mutations which enhance NB1 and MSD1 interaction in F508del CFTR cells, thus indicating that a combination of correctors, which bind to alternative locations on CFTR, may have a synergistic effect in improving CFTR function.
However in vivo results from Clancy et al. regarding the benefit of oral lumacaftor in F508del homozygous adult patients were found to be inconclusive.[Citation42] Although CFTR chloride conductance was significantly improved, as shown by reductions in sweat chloride concentrations, lung function was not enhanced. The short nature of this study and the positive effects on sweat chloride levels warrants nevertheless supplementary, long-term research.
VX-661
Further CFTR correctors have been identified, including the recently developed VX-661 – a close chemical analog of VX-809 and with an anticipated identical mechanism of action. Initial reports from VX-661 studies appearing promising.[Citation43] CFTR function was significantly improved, as specified by the increased chloride transport in F508del mutant cells in vitro and in vivo research identified decreased sweat chloride levels and improved FEV1 in patients who are homozygous for F508del mutation subsequent to VX-661 administration alone or in combination with ivacaftor. Initial reports confirm that this combination of treatments actually improves pulmonary function in those patients who are F508del/G551D heterozygous, thus expanding the proportion of CF patients who may benefit from CFTR modulator therapy.[Citation44]
Combination of corrector and potentiator therapy
A corrector, which enhances the amount of mature CFTR at the cell surface, in combination with a potentiator, which acts to increase the conductance of these channels, is hypothesized as a treatment technique in patients with the F508del mutation. Indeed, application of the potentiator ivacaftor further enhances the channel opening of CFTR HBE cells which have been initially corrected with lumacaftor in vitro, with chloride transport to the epithelial surface also increasing.[Citation40]
A recent global clinical trial has examined the effects of the administration of lumacaftor in combination with ivacaftor in adult patients.[Citation45] Significant improvements in sweat chloride levels were noted in cohort one, all of whom were F508del homozygous. Cohorts two and three included both homozygous and heterozygous F508del CF patients. Combination therapy in this subpopulation did not significantly reduce sweat chloride levels, yet strangely, significant improvements in FEV1 were detected. Further sub-analysis of these results established that there was no substantial increase in the FEV1 of F508 heterozygous participants, most likely because of the reduced levels of F508del CFTR available to correct and subsequently potentiate. These results suggest that ivacaftor and lumacaftor combination therapy may confer a clinical benefit to those who are homozygous for the F508del CFTR mutation. The further development of two ongoing, large phase-III trials, TRAFFIC and TRANSPORT, focus on the efficacy and safety of combination therapy in these homozygous patients.
Results have been published by Vertex Pharmaceuticals for TRAFFIC and TRANSPORT Trials, with modest but significant increases in FEV1, less pronounced than the effect of ivacaftor on G551D patients, being described following treatment with Orkambi (dual ivacaftor and lumacaftor treatment).[Citation46] Along with the observation that the regimen was well tolerated, there were also reduced numbers of pulmonary exacerbations, with the successful outcomes of this study leading to FDA approval. The beneficial effect of lumacaftor and ivacaftor combination therapy in F508del homozygous patients has yet to be fully established, as no significant improvements in patient reported symptoms have been recorded. This may be because of the research being in its early stages, a limiting factor which will be overcome following final publication. Interim outcomes demonstrated that dual treatment for up to 48 weeks maintained improvements in FEV1. Therefore, it appears that for F508del homozygotes, dual treatment results in increased FEV1, have an acceptable side effect profile, reduced exacerbations and hospital admissions.[Citation46] Nevertheless, even considering these promising clinical outcomes, lumacaftor–ivacaftor dual therapy remains a costly treatment, with estimates of up to US$ 300,000 per year in some countries.[Citation47]
Dual acting molecules
An interesting progression in CFTR targeting is the detection of dual acting molecules, which act to both correct and potentiate the mutated F508del CFTR. Sheer serendipity led to the discovery of trimethylangelicin (TMA), an anti-inflammatory IL-8 inhibitor which also acts to potentiate and correct F508del CFTR in FRT cells.[Citation48] The dual-action of TMA should be established confirmed in HBE cells, which have a superior physiological relevance in comparison to FRT cells.
The fusion of the active components of a potentiator and corrector is an additional technique that could be utilized to create a one-step, dual acting compound able to target F508del mutated CFTR. The development of this hybrid molecule was achieved by Mills et al.[Citation49] This proof-of-concept study demonstrated that the dual-acting molecule can be cleaved by intestinal enzymes to create the two active compounds responsible for corrector and potentiator action. Dual acting molecules are at preliminary stages in their development and research, and it is unclear if dual molecules would provide an advantage, other than ease of use, in comparison with monotherapy.
Gene therapy strategies for treating CF
Unlike treatment with CFTR modulators, an attractive quality of gene therapy is that it could act to correct all CFTR mutations, regardless of class. Viral vectors exhibiting a natural propensity for the respiratory epithelium, such as adenovirus, were initially anticipated to be ideal candidates for the delivery of DNA into target cells. Indeed, the initial administration of an adenoviral vector housing functional CFTR cDNA resulted in the successful expression of the CFTR protein in some airway epithelial cells.[Citation50] Expression of CFTR was, however, unsuccessful in the majority of patients enrolled in this study, a finding possibly attributed to variable host defense mechanisms. Supporting this, the majority of cells recovered on sampling of the airways were inflammatory cells.
Subsequent studies questioned the use of adenoviral vectors, following the discovery that 3-monthly repeated administration of the vector resulted in diminished CFTR expression and the development of anti-adenoviral antibodies.[Citation51]
Alternative viral vectors that do not induce an immune response such as adeno-associated viruses (AAVs) were suggested as an alternative, primarily because of their lack of pathogenicity in humans. Nevertheless, research has not demonstrated a clear clinical advantage for AAVs use as a vector for gene therapy in CF.[Citation52] Indeed, clinical trials have not demonstrated a clear benefit for the use of AAV vectors. A randomized trial involving 102 participants with mild to moderate CF were randomized to receive two doses of either a functioning CFTR through an AAV vector or a placebo, both given by means of an aerosol.[Citation53] One month after administration, there were no significant differences observed between treatment and placebo groups. In addition, markers of inflammation and antibiotic use were similar between groups.
Non-viral vectors for the delivery of CFTR DNA were created by complexing DNA with lipids, peptides and other molecules. A fourth generation plasmid, pGM169, which lacks the CpG islands thought to initiate the inflammatory and immune response, has been shown to successfully deliver CFTR DNA into cells and this resulted in persistent CFTR expression in the mouse.[Citation54] A more recent study explored the use of nebulized liposome-mediated gene delivery into the airway epithelium.[Citation55] This phase I/IIa study entailed the delivery of a DNA plasmid encoding CFTR into the airways and led to the conclusion that repeated prolonged administration would be required for future trials.
An alternative method of DNA delivery is using human artificial chromosomes (HAC). The HAC contains a larger version of the CFTR gene in comparison with the typical cDNA used in viral vectors and behaves as an additional cellular chromosome. The delivery of a complete CFTR gene, including regulatory and promoter regions, has been suggested as a method of enhancing gene expression and preventing gene silencing which often accompanies vector delivery of cDNA. Studies by Rocchi et al. have demonstrated the ability of bacteria to insert the HAC with functional CFTR into mammalian cells and maintain gene expression over 50 cell generations.[Citation56]
The discovery and characterization of CRISPR/Cas9 for correcting mutated genes
The notion of RNA-directed genome editing was founded upon the discovery of clustered regularly interspaced short palindromic repeats (CRISPRs), originally observed in Escherichia coli in 1987.[Citation57] Further elaborations determined CRISPR loci presence within both prokaryote plasmid and chromosomal DNA (cDNA), comprising an array of short, identical repeat sequences delineated by unique genome-targeting spacers.[Citation58] It was further noted that these CRISPR loci frequently featured alongside a series of CRISPR-associated (Cas) genes of high sequence homology to DNA repair enzymes.
Revelation of the CRISPR and Cas collective mechanism and its precise role within prokaryotic adaptive immunity was achieved through studies undertaken by Garneau et al. regarding the Streptococcus thermophiles bacterium.[Citation59] Deltcheva et al. [Citation60] later elaborated the now widely described and frequently studied Streptococcus pyogenes type II CRISPR-Cas9 defense system. Within these studies, it was shown that initial acquisition of CRISPR-Cas9 bacterial immunological memory occurs within an adaptation phase, whereby DNA spacers are derived from foreign, invading nucleic acid sequences and subsequently stored within the CRISPR array. This memory may then be accessed and employed within prospective expression and interference phases, to affect the recognition and destruction of previously encountered pathogens.
Throughout this expression phase, established CRISPR sequences are transcribed and cleaved enzymatically to form CRISPR ribonucleic acid (crRNA) molecules, derived from and therefore capable of pairing with complementary sequences of previously encountered foreign DNA. crRNA is subsequently hybridized with trans-activating crRNA (tracrRNA) through the action of Cas proteins and accessory factors. This activity generates a mature, dual-RNA crRNA-tracrRNA structure, which may then associate with and guide the double-stranded DNA endonuclease enzyme, Cas9 (alternatively called Cas5, Csn1 or Csx12).[Citation61] Cas9 contains two domains, HNH nuclease and RuvC-like, which are important within the interference phase in the cleavage of complementary and non-complementary DNA strands respectively to cause a double-stranded break (DSB).[Citation62] In this way, CRISPR-Cas9 complexes specifically target and degrade mobile genetic elements at sites complementary to the crRNA sequence, providing a protective measure against invading pathogens.
CRISPR-Cas9’s value as a biomedical tool is furthermore enhanced by the diversity of its potential outcomes, as it operates in tandem with innate cellular repair pathways. Two distinct repair mechanisms may be invoked following recognition of DNA damage. Either non-homologous end-joining (NHEJ) or homologous recombination (HR) may be initiated as a method of DNA repair, with two divergent outcomes.
NHEJ pertains to the intrinsic chemical ligation of non-homologous ends of DNA. NHEJ serves as the preferential pathway, should a DSB be affected in the absence of a repair template.[Citation63] NHEJ is inherently more error-prone, commonly imposing small-scale insertions or deletions (indel mutations) at the DSB site.[Citation64] Such activity presents the possibility of producing frame-shift DNA mutations. These are particularly problematic if positioned within an exon, as they pose risk of detrimental phenotypic effects caused by the improper transcription and translation of genes into proteins. As such, within the laboratory, the delivery of isolated CRISPR-Cas9 independent of a donor repair template can be manipulated to facilitate gene knock-out.
CRISPR-Cas9 furthermore presents the opportunity to perform knock-in interventions as a feature of new DNA repair therapies. This knock-in functionality is conversely facilitated by the HR pathway, whereby an extra-chromosomal donor DNA fragment may be delivered alongside the CRISPR-Cas9 editing system for chromosomal integration in place of the excised region.[Citation65] Repair templates necessarily consist of the corrected gene sequence, flanked by sequences with homology both up and downstream of the DSB.[Citation66] This technique can be used to both correct small point mutations and insert longer, functional gene fragments [Citation67] and has subsequently been proposed within potential therapies spanning the full spectrum of genetic conditions, including CF.
The fundamental value of CRISPR-Cas9 technology across the domains of biology, biotechnology and therapeutic biomedicine lies in the inherent simplicity of the system’s ‘reprogrammability’. This feature arises from the discrete nature of the two main elements responsible for nuclease cleavage and DNA binding in that the generation of new and specifically targeted molecules requires the synthesis of a short DNA binding domain in isolation.[Citation63] This is in contrast to the intensive and sophisticated protein engineering techniques required for the adaptation of modular site-specific proteins in the recognition of new targets, including ZNF and TALEN.[Citation68] The simple modification of oligonucleotide protein-binding domains required in the extension of CRISPR-Cas9 system functionality has proven to be simpler, more cost-effective and necessitating only basic biologic training, thereby improving methodological availability. CRISPR-Cas9 technology could therefore theoretically be applied to induce genomic changes at any site across the human genome with a neighboring 5ʹ-NGG protospacer-adjacent motif.
Improvements to the wild-type Cas9 nuclease are also under way, to permit enhanced expression and activity in human cell models. Studies to date have mutated the wild-type Cas9 nuclease enzyme to generate a Cas9 DNA nickase through the modification of the RuvC I domain.[Citation69] Mutated Cas9 nickase activity is limited to the cleavage of a single DNA strand, in similar fashion to TALEN and ZFN technologies. Mutated Cas9 is thereby employed in a ‘double-nicking strategy’, whereby two Cas9 molecules are paired with two distinct RNA molecules and then used in tandem to target a single site within the genome.[Citation70] ‘Double-nicking’ has the advantage of improving site specificity, as it requires the simultaneous identification of two genomic regions to affect a single DSB. Furthermore, double-nicking encourages seamless base-excision (or high-fidelity HR) DNA repair and thus avoids potentially detrimental on- and off-target NHEJ. Important benefits of this manipulative measure can be seen in practice, through reduced frequency of indel in off-target locations.[Citation71]
Advantages of using targeted nucleases such as CRISPR/Cas9 for treating CF
Within the an adult stem cell gene therapy study conducted by Schwank et al. [Citation8], CRISPR-Cas9 was used to correct F508 del CFTR mutations in adult intestinal stem cells isolated from two homozygous, pediatric CF patients. These cells were subsequently cultured in vitro to form intestinal organoids, representing a three-dimensional small intestinal epithelium. The organoids were transfected with donor plasmids encoding both a wild-type CFTR donor sequence and CRISPR-Cas9, with the purpose of affecting HR directed genetic mutagenesis. Within the CRISPR-Cas9 system, two distinct guides RNA were delivered, targeted at either CFTR exon 11 or intron 11. Subsequent treatment with a forskolin-induced swelling assay indicated the presence of functional CFTR comparable with wild-type organoids. Furthermore, analysis of transgenic clone cDNA revealed site-specific integration of the donor sequence, indicating successful genotypic repair. It should be noted however, that a puromycin resistance selection procedure was used to obtain correctly gene-edited cells, meaning the efficiency of the procedure appears relatively low. This approach was an extension of similar studies using mouse tissue, whereby the intestine products were successfully grafted into the mouse hosts.[Citation72] Safety reports were also favorable, with only a single off-target mutation detected within an intron, thereby presenting a low probability of detrimental phenotypic consequences.
The authors of this study furthermore concluded that the failure of this technique to address CF lung-related pathophysiology is likely to contraindicate its value when applied to the context of clinical setting. However, it was also suggested that such organoid technology may in future complement iPSC-based gene correction approaches, which have been proposed for use within potential therapeutic applications for CF patients with lung disease.[Citation8]
iPSC-based approaches to mutant CFTR correction have been achieved previously using ZFN gene-editing technology [Citation73] and more recently been demonstrated successfully using CRISPR.[Citation74] In this study, donor fibroblasts were isolated from a CF patient skin biopsy for reprogramming to iPSCs. CF iPSCs were then co-nucleofected with a Cas9 vector, CFTR gRNA vector and donor vector in various combinations. Integration analysis pertaining to 36 clones revealed a 16.7% efficiency and further genetic sequencing analysis confirmed the presence of the corrected CFTR gene. Upon further genetic analysis, no mutations were found within 300 bp of predicted off-target sites and random integration of components was lacking elsewhere in the genome. Gene-corrected CF iPSC cell lines were subsequently differentiated into proximal airway epithelial cells. The functional propensity of these cells was then tested using CFTR current analysis through whole-cell patch clamp technique. Results revealed CFTR conductivity similar to wild-type iPSC derived lung epithelial cells. Furthermore, CFTR present was found capable of undergoing the conformational changes (N-glycosylation) necessary for cell membrane translocation.
Positive results have also been obtained when attempting the precise correction of the dystrophin gene in patient stem cells using TALEN and CRISPR-Cas9 by using an exon knock-in approach.[Citation75]
Obstacles preventing the use of targeted nucleases for treating CF
While the proficient function of CRISPR-Cas9 technology has been proven to date, safety and efficacy concerns remain. Off-target and unintended on-target mutations remains a significant and major obstacle for the technology’s extension and application to therapeutic genome editing.
Off-target mutations are errors occurring because of imperfect guide RNA specificity and the subsequent pairing of the CRISPR-Cas9 complex at cDNA sites other than the intended target gene. Should this occur, evocation of NHEJ repair mechanisms at these sites may result in unanticipated and potentially harmful mutations. Off-target mutation rates are believed to be concentration-dependent, occurring more frequently with increased DNA transfection quantity and may therefore be avoided with careful titration of enzyme and RNA dosage.[Citation76] Advancements in specificity and thus reduction of off-target mutagenesis has been observed through the use of multiple RNA-Cas9 complexes in double-nicking strategies [Citation71] and improved natural Cas orthologues, alongside careful selection of target RNA sequences [Citation77] and adaptations of guide RNA molecules.[Citation78] However, a recent study by Wang et al. revealed a high frequency of off-target single-base bulges as the result of CRISPR-Cas9 transfection, with up to 13 guide RNA mismatches.[Citation79]
Further safety concerns are elicited by the potential risk of permanent integration of CRISPR-Cas9 components into the host genome following viral vector transfection methods.[Citation75] It is thought that the continued production of these components beyond the desired period may further increase the risk of off-target events. Counterarguments suggest, however, that the risks associated with permanent CRISPR-Cas9 genomic integration are insignificant owing to an absence of reports of instability within subsequent generations of cultured cell lines and, furthermore, the retained ability for knock-in murine models to breed naturally over the course of multiple generations in the absence of perceived tissue, reproductive or health abnormalities [Citation80].
Undesirable on-target mutation is a further concern. On-target mutation is made more likely upon insufficient donor template availability as error-prone NHEJ repair mechanisms are favored over HR.[Citation81] Failure to provide excessive quantities of a repair template is of particular concern in cell lines homozygous for the target mutant gene. Within these cells, the relative abundance of uncleaved alleles increases the likelihood of their preferential utilization over the donor template within HR repair mechanisms, unless provided in excess.[Citation82] To this affect, the yield of correctly edited alleles is reduced. Further strategies are also required to lower NHEJ and raise HR to reduce the frequency of unintended on-target indel errors.
The necessity of computationally identifying potential off-target binding sites prior to transfection has been stressed, as a means of improving the safety and efficacy of CRISPR-Cas9 methodology. However, even using intensive in silico methods of identification of all sites with prominent sequence homology does not provide an absolute and predictable indication of off-target mutation frequency, as Cas9 potential reactivity is unpredictable.[Citation83] This necessitates empirical observation to detail the actual genomic sites highly-bound by Cas9 and the sequencing of altered genetic products to detect unintended outcomes. Presently standardized parameters for measuring off-target effects following successful genetic mutagenesis are lacking.
Conclusions
Despite being in its early stages, the use of the CRISPR-Cas9 system is very promising considering the proof that the faulty CFTR gene can be corrected in stem cells. This opens many avenues for the precise treatment of CF. Target organ-directed treatment strategies such as direct delivery of the therapeutic agents into the airways or alternately the generation of CFTR-corrected stem respiratory epithelial cells to be subsequently administered to the patient should address the potential ethical concerns related to the potential impact of targeted nucleases upon germline cells. However, studies to date have consistently failed to achieve total repair efficiency in all transfected cells, suggesting the need for additional research to elucidate the processes responsible for the repair of cleavage sites and to improve yield.
Another obstacle that needs overcoming is designing an efficient delivery vehicle suitable for targeting airways epithelium in vivo. Adeno-associated viruses are the most used vehicles for CRISPR delivery in vivo. Nevertheless, lipid or polymer nanoparticles may avoid some of the viral vector disadvantages as mentioned by Li et al.[Citation84] The magnitude of the CRISPR-Cas9 complex complicates processes using lentiviral and adeno-associated viral vectors [Citation85,Citation86] in addition to the clinical limitations inherent in larger capacity viral transfection methods.[Citation75] Identifying and targeting self-renewing human lung stem cell populations such as bronchioalveolar stem cells, Clara cells and alveolar type II progenitors is proposed as a more effective means of supporting continued CFTR production post-treatment.[Citation87] Therefore improved methods must be devised to improve CRISPR complex patient administration.
Nevertheless, using CRISPR to correct gene defects is more practical and poses less potential subsequent problems then ‘classical’ gene therapy where the correct gene could potentially be inserted elsewhere in the genome and eventually cause, for example, hematologic malignancies, if inserted in proximity to certain promoters. By correcting the disease gene ‘in situ’ CRISPR maintains its location in relation with regulatory components and adjacent genomic structures. From this viewpoint, CRISPR could be seen as an improved form of gene therapy rather than a distinct, entirely different type of treatment strategy.
The significant promise carried out by CRISPR-based therapies is underlined by the recent in vivo work published by Ding et al. in 2014 [Citation88] in which the authors describe the permanent alteration of the PCSK9 gene in mice using CRISPR. The authors achieved mutagenesis of around 50% of the PCSK9 gene in the liver 3–4 days after a single CRISPR administration without any off- target mutagenesis detectable at 10 selected sites. Two very recent publications [Citation6,Citation7] related to the application of CRISPR for treating muscular dystrophies further confirm the promise of this new technology for treating genetic diseases. Therefore, whilst overcoming the obstacles related to the accessibility of the airways and indeed improvements in efficacy are needed, it is not unrealistic to expect it have a potential use for treating cystic fibrosis.
Expert commentary
The genotyping and classification of CFTR mutations led to the discovery of precise treatments that specifically correct certain mutations. In this respect, the introduction of new drugs such as ivacaftor and lumacaftor represent major advances for CF treatment that are expected to increase the life expectation of a significant number of patients. Furthermore, the recent discovery of CRISPR and the proof-of-concept correction of a defective CFTR gene from a CF patient in a stem cell organoid open the perspective of establishing a precise, personalized CF treatment that could reverse the genetic lesion in these patients.
Five-year view
While promising, CRISPR-Cas9 remains a nascent technology and thus requires careful scrutiny over concerns of efficacy and safety prior to translation into the clinical environment. At present, two major concerns remain imperfect repair efficiency and specificity. Crucially, means of preventing Cas9 off-target activity must be devised. Additional evaluation of long-term biological consequences is also important to ensure a lack of toxicity and side effects throughout prolonged periods of exposure.[Citation89]
Key issues
Around 2000 mutations have been identified in ~70,000 patients worldwide as causative of CF.
Mutation classification led to precise treatments of CF using mutation specific medications.
Gene therapies aim to generate functionally active CFTR proteins in respiratory epithelial cells.
The discovery of CRISPR (Classes of Regularly Interspaced Palindromic Repeats) and Cas9 (Crispr-ASsociated) nuclease system opened the perspective of specifically correcting defective CFTR.
Difficulties of delivering the nucleases into the target respiratory epithelium cells represent a major problem to be resolved.
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
- Fanen P, Wohlhuter-Haddad A, Hinzpeter A. Genetics of cystic fibrosis: CFTR mutation classifications toward genotype-based CF therapies. Int J Biochemistry Biol. 2014;52:94–102.
- Collaco JM, Blackman SM, McGready J, et al. Quantification of the relative contribution of environmental and genetic factors to variation in cystic fibrosis lung function. J Pediatr. 2010;157:802–823.
- Amaral MD, Balch WE. Hallmarks of therapeutic management of the cystic fibrosis functional landscape. J Cyst Fibros. 2015;14(6):687–699.
- Durmowicz AG, Witzmann KA, Rosebraugh CJ, et al. Change in sweat chloride as a clinical end point in cystic fibrosis clinical trials: the ivacaftor experience. CHEST J. 2013;143(1):14–18.
- Cutting GR. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet. 2014;16(1):45–56.
- Tabebordbar M, Zhu K, Cheng JKW, et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science [Internet]. 2015 Dec 31 [cited 2016 Jan 10]; Available from: http://www.sciencemag.org/cgi/doi/10.1126/science.aad5177.
- Nelson CE, Hakim CH, Ousterout DG, et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science [Internet]. 2015 Dec 31 [cited 2016 Jan 10]; Available from: http://www.sciencemag.org/cgi/doi/10.1126/science.aad5143.
- Schwank G, Koo B-K, Sasselli V, et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 2013;13(6):653–658.
- Ikpa PT, Bijvelds MJC, de Jonge HR. Cystic fibrosis: toward personalized therapies. Int J Biochem Cell Biol. 2014;52:192–200.
- Chen J, Cai Z, Li H, et al. Function of CFTR protein: ion transport. Prog Respir Res. 2006;34:38–44.
- Quinton PM. Cystic fibrosis: impaired bicarbonate secretion and mucoviscidosis. Lancet. 2008 Aug;372(9636):415–417.
- Derichs N, Jin B-J, Song Y, et al. Hyperviscous airway periciliary and mucous liquid layers in cystic fibrosis measured by confocal fluorescence photobleaching. FASEB J. 2011;25:2325–2332.
- Rowe SM, Accurso F, Clancy JP. Detection of cystic fibrosis transmembrane conductance regulator activity in early-phase clinical trials. Proc Am Thorac Soc. 2007;4:387–398.
- McKone EF, Velentgas P, Swenson AJ, et al. Association of sweat chloride concentration at time of diagnosis and CFTR genotype with mortality and cystic fibrosis phenotype. J Cyst Fibros. 2015;14(5):580–586.
- Dodge JA, Lewis PA, Stanton M, et al. Cystic fibrosis mortality and survival in the UK: 1947-2003. Eur Respir J. 2007;29(3):522–526.
- CFTR2 TCaFToC [Internet]. 2015 [cited 2016 Feb 2]. Available from: http://www.cftr2.org/
- Rowntree RK, Harris A. The phenotypic consequences of CFTR mutations. Ann Hum Genet. 2003;67:471–485.
- Du K, Sharma M, Lukacs GL. The ΔF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat Struct Mol Biol. 2005;12:17–25.
- Bronsveld I, Mekus F, Bijman J, et al. Chloride conductance and genetic background modulate the cystic fibrosis phenotype of ΔF508 homozygous twins and siblings. J Clin Invest. 2001;108:1705–1715.
- Howell LD, Borchardt R, Cohn JA. ATP hydrolysis by a CFTR domain: pharmacology and effects of G551D mutation. Biochem Biophys Res Commun. 2000;271(2):518–525.
- Denning GM, Ostedgaard LS, Welsh MJ. Abnormal localization of cystic fibrosis transmembrane conductance regulator in primary cultures of cystic fibrosis airway epithelia. J Cell Biol. 1992;118:551–559.
- Lukacs GL, Chang XB, Bear C, et al. The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J Biol Chem. 1990;268:21592–21598.
- Howard M, Frizzell RA, Bedwell DM. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat Med. 1996;2:467–469.
- Zhang L, Button B, Gabriel SE, et al. CFTR delivery to 25% of surface epithelial cells restores normal rates of mucus transport to human cystic fibrosis airway epithelium. PLoS Biol. 2009;7:e1000155.
- Sermet-Gaudelus I, Renouil M, Fajac A, et al. In vitro prediction of stop-codon suppression by intravenous gentamicin in patients with cystic fibrosis: a pilot study. BMC Med. 2007;5:5.
- Clancy JP, Rowe SM, Bebok Z, et al. No detectable improvements in cystic fibrosis transmembrane conductance regulator by nasal aminoglycosides in patients with cystic fibrosis with stop mutations. Am J Respir Cell Mol Biol. 2007;37:57–66.
- Welch EM, Barton ER, Zhuo J, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91.
- Wilschanski M, Miller LL, Shoseyov D, et al. Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis. Eur Respir J. 2011;38:59–69.
- Kerem E, Konstan MW, De Boeck K, et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir Med. 2014;2:539–547.
- Gonzalez-Hilarion S, Beghyn T, Jia J, et al. Rescue of nonsense mutations by amlexanox in human cells. Orphanet J Rare Dis. 2012;7:58–58.
- Van Goor F, Hadida S, Grootenhuis PDJ, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A. 2009;106:18825–18830.
- Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663–1672.
- Davies JC, Wainwright CE, Canny GJ, et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med. 2013;187:1219–1225.
- Moss RB, Flume PA, Elborn JS, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: a double-blind, randomised controlled trial. Lancet Respir Med. 2015;3(7):524–533.
- McKone EF, Borowitz D, Drevinek P, et al. Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp-CFTR mutation: a phase 3, open-label extension study (PERSIST). Lancet Respir Med. 2014;2:902–910.
- Yeh H-I, Yeh J-T, Hwang T-C. Modulation of CFTR gating by permeant ions. J Gen Physiol. 2015;145:47–60.
- Bompadre SG, Li M, Hwang T-C. Mechanism of G551D-CFTR (cystic fibrosis transmembrane conductance regulator) potentiation by a high affinity ATP analog. J Biol Chem. 2008;283:5364–5369.
- Flume PA, Liou TG, Borowitz DS, et al. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. CHEST J. 2012;142:718–724.
- Pedemonte N, Lukacs GL, Du K, et al. Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J Clin Invest. 2005;115:2564–2571.
- Van Goor F, Hadida S, Grootenhuis PD, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A. 2011;108:18843–18848.
- Ren HY, Grove DE, De La Rosa O, et al. VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Mol Biol Cell. 2013;24(19):3016–3024.
- Clancy JP, Rowe SM, Accurso FJ, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax. 2012;67:12–18.
- Donaldson S, Pilewski J, Griese M, et al. WS7.3 VX-661, an investigational CFTR corrector, in combination with ivacaftor, a CFTR potentiator, in patients with CF and homozygous for the F508Del-CFTR mutation: interim analysis. J Cyst Fibros. 2013;12(Supplement 1):S14.
- Vertex [Internet]. 2014 [cited 2016 Feb 2]. Available from: http://investors.vrtx.com/releasedetail.cfm?ReleaseID=844677
- Boyle MP, Bell SC, Konstan MW, et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med. 2014;2:527–538.
- Wainwright CE, Elborn JS, Ramsey BW, et al. Lumacaftor–ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. New England J Med. 2015;373(3):220–231.
- Rehman, et al. Lumacaftor–ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. New England J Med. 2015;373(18):1783–1784.
- Favia M, Mancini MT, Bezzerri V, et al. Trimethylangelicin promotes the functional rescue of mutant F508del CFTR protein in cystic fibrosis airway cells. Am J Physiol Lung Cell Mol Physiol. 2014;307:L48–61.
- Mills AD, Yoo C, Butler JD, et al. Design and synthesis of a hybrid potentiator-corrector agonist of the cystic fibrosis mutant protein DeltaF508-CFTR. Bioorg Med Chem Lett. 2010;20:87–91.
- Crystal RG, McElvaney NG, Rosenfeld MA, et al. Administration of an adenovirus containing the human CFTR cDNA to the respiratory tract of individuals with cystic fibrosis. Nat Genet. 1994;8:42–51.
- Harvey BG, Leopold PL, Hackett NR, et al. Airway epithelial CFTR mRNA expression in cystic fibrosis patients after repetitive administration of a recombinant adenovirus. J Clin Invest. 1999;104:1245–1255.
- Griesenbach U, Alton EW. Progress in gene and cell therapy for cystic fibrosis lung disease. Curr Pharm Des. 2012;18:642–662.
- Moss RB, Milla C, Colombo J, et al. Repeated aerosolized AAV-CFTR for treatment of cystic fibrosis: a randomized placebo-controlled phase 2B trial. Hum Gene Ther. 2007;18:726–732.
- Hyde SC, Pringle IA, Abdullah S, et al. CpG-free plasmids confer reduced inflammation and sustained pulmonary gene expression. Nat Biotechnol. 2008;26:549–551.
- Alton EWFW, Boyd AC, Porteous DJ, et al. A Phase I/IIa safety and efficacy study of nebulized liposome-mediated gene therapy for cystic fibrosis supports a multidose trial. Am J Respir Crit Care Med. 2015;192(11):1389–1392.
- Rocchi L, Braz C, Cattani S, et al. Escherichia coli-cloned CFTR loci relevant for human artificial chromosome therapy. Hum Gene Ther. 2010;21:1077–1092.
- Ishino Y, Shinagawa H, Makino K, et al. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol. 1987;169(12):5429–5433.
- Barrangou R, Fremaux C, Deveau H, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315(5819):1709–1712.
- Garneau JE, Dupuis M-È, Villion M, et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468(7320):67–71.
- Deltcheva E, Chylinski K, Sharma CM, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471(7340):602–607.
- Rath D, Amlinger L, Rath A, et al. The CRISPR-Cas immune system: biology, mechanisms and applications. Biochimie. 2015;117:119–128.
- Nishimasu H, Ran FA, Hsu PD, et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156(5):935–949.
- Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–2308.
- Jackson SP, Khanna K. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27(3):247–254.
- Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–826.
- Heyer W-D, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. Annu Rev Genet. 2010;44(1):113–139.
- Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014 Mar 2;32(4):347–355.
- Van Erp PB, Bloomer G, Wilkinson R, et al. The history and market impact of CRISPR RNA-guided nucleases. Curr Opin Virol. 2015;12:85–90.
- Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823.
- Ran FA, Hsu PD, Lin C-Y, et al. Double Nicking by RNA-Guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154(6):1380–1389.
- Shen B, Zhang W, Zhang J, et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods. 2014;11(4):399–402.
- Yui S, Nakamura T, Sato T, et al. Functional engraftment of colon epithelium expanded in vitro from a single adult Lgr5+ stem cell. Nat Med. 2012;18(4):618–623.
- Crane AM, Kramer P, Bui JH, et al. Targeted correction and restored function of the CFTR gene in cystic fibrosis induced pluripotent stem cells. Stem Cell Reports. 2015;4(4):569–577.
- Firth AL, Menon T, Parker GS, et al. Functional gene correction for cystic fibrosis in lung epithelial cells generated from patient iPSCs. Cell Rep. 2015;12(9):1385–1390.
- Lisa Li H, Nakano T, Hotta A. Genetic correction using engineered nucleases for gene therapy applications. Dev Growth Differ. 2014;56(1):63–77.
- Hsu PD, Scott DA, Weinstein JA, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31(9):827–832.
- Cho SW, Kim S, Kim Y, et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014 Jan 1;24(1):132–141.
- Fu Y, Sander JD, Reyon D, et al. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32(3):279–284.
- Wang X, Wang Y, Wu X, et al. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat Biotechnol. 2015;33(2):175–178.
- Parikh BA, Beckman DL, Patel SJ, et al. Detailed phenotypic and molecular analyses of genetically modified mice generated by CRISPR-Cas9-mediated editing. Plos One. 2014;10(1):e0116484.
- Cox DBT, Platt RJ, Zhang F. Therapeutic genome editing: prospects and challenges. Nat Med. 2015;21(2):121–131.
- Bassett AR, Liu J-L. CRISPR/Cas9 and genome editing in drosophila. J Genet Genomics. 2014;41(1):7–19.
- Wu X, Scott DA, Kriz AJ, et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol. 2014;32(7):670–676.
- Li L, He Z-Y, Wei X-W, et al. Challenges in CRISPR/CAS9 delivery: potential roles of nonviral vectors. Hum Gene Ther. 2015;26(7):452–462.
- Kumar M, Keller B, Makalou N, et al. Systematic determination of the packaging limit of lentiviral vectors. Hum Gene Ther. 2001;12(15):1893–1905.
- Wu Z, Yang H, Colosi P. Effect of genome size on AAV vector packaging. Mol Ther. 2010;18(1):80–86.
- Kajstura J, Rota M, Hall SR, et al. Evidence for human lung stem cells. New England J Med. 2011;364(19):1795–1806.
- Ding Q, Strong A, Patel KM, et al. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ Res. 2014;115:488–492.
- Barrangou R, May AP. Unraveling the potential of CRISPR-Cas9 for gene therapy. Expert Opin Biol Ther. 2015;15(3):311–314.