
Many efforts are being made to exploit progress in the molecular understanding of disease pathology and the availability of biomarkers to advance modern medical practice. In the individualized therapeutic approach of modern precision medicine, treatment should no longer be directed against a monolithic disease entity, but rather against specific features of the pathology of the individual patient. From a public health perspective, precision medicine may contribute to avoiding side effects, drug failures, and market recalls. Biomarker tests are available in today’s clinical practice that allow clinicians to increase the specificity of diagnosis, individually choose therapy and doses, as well as estimate the individual safety risk [Citation1]. Molecular approaches potentially enable clinicians to identify disease subgroups that may profit from different treatment strategies. Since the assessment of benefit/risk ratios constitutes a key issue in regulatory decisions, the integration of information from biomarkers on this assessment represents a challenge to regulatory work. Hence, the objective of modern regulation is to achieve a stratified risk–benefit assessment in considering biomarker evidence [Citation2].
In drug regulation, the term companion diagnostics has been defined as an in vitro diagnostic device that could be essential for the safe and effective use of a corresponding therapeutic product [Citation3]. As is stated in the guideline, labeling of a therapeutic product lists the conditions when use of a diagnostic device is required. A distinction is made between uses of diagnostic devices that are suggested but not required (also named as complementary diagnostics) and obligatory diagnostics considered ‘essential.’ Regulatory advice often addresses the question when to include biomarker-negative patients as compared to the biomarker-positive in drug development trials, or guidance on the issuing of warning information on biomarker-defined patient subgroups in the drug labels. Regulatory decisions such as these require evidence on the validity of a biomarker test in specific conditions. Similarly, clinicians and health care professionals applying stratified treatment approaches need to understand when and to whom the biomarker test may be applied.
Therefore, an important prerequisite for the rational use of biomarkers as companion diagnostics is evidence about their generalizability across drugs, patient groups, diseases, and ethnicities. Issues of generalizability have always been a primary concern of regulators, determining what medication uses are in-label and what are off-label. Drug labels describe the situations for which there is enough evidence for a positive benefit/risk ratio in the use of the drug. The same critical concern should be applied to biomarkers with respect to their applicability as companion diagnostics. illustrates the boundaries to the generalizability of biomarkers used as companion diagnostics focusing on the field of drug safety, drug metabolism, drug response, and response surveillance.
Figure 1. Biomarker testing for companion diagnostics.
Different degrees of generalizability of findings from biomarker testing: green light (widely generalizable results), yellow light (caution is warranted), and red light (likely difficulties in generalizing outcomes to other contexts). The generalization domains are displayed in the column headings, while the row names refer to the biomarker classes.
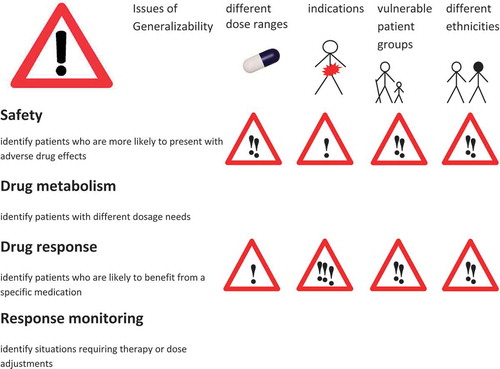
Safety biomarkers mostly consist of genetic germline variants and provide information that is usually generalizable across different disease indications requiring the same therapy. However, the absolute amount of dose or drug exposure affects the individual safety risk. Therefore, care should be given to ensure that the dose range in which the drug is given does not invalidate the biomarker predictive value.
Safety biomarkers generally maintain their predictive validity for all patients who are exposed to the drug and will generally deliver reliable information in vulnerable patient populations as well as in different ethnicities. However, the ‘attention sign’ to generalizability across patient populations of considers the existence of additional clinical parameters or risk factors that may influence the net predictive validity of the biomarkers. As an example, the human leukocyte antigene (HLA) genotype HLA-B*1502 predicting the risk of carbamazepine-associated hypersensitivity reaction is prevalent in certain Asian populations and is labeled as a required or recommended companion diagnostic biomarker in USA and Canada, whereas in Europe where the allele is largely absent, the label does not contain biomarker information or recommendations for testing [Citation4]. The majority of biomarkers currently mentioned in drug labels belong to this group, addressing issues of drug safety, either directly or indirectly via drug interactions or metabolism. The advantage of presenting biomarker information in drug labels is that it provides information that may be used for an individualized patient treatment strategy while maintaining the in-label use of the drug. Even if valid across different drug indications and in principle the most generalizable biomarkers, safety biomarkers are often first tested after side effects have occurred. In contrast, a precision medicine-inspired approach would promote broad testing of generally applicable biomarkers for drug safety according to the well-known injunction: first do no harm.
Drug metabolizing enzyme biomarkers may be used for dose adjustments in accordance to the metabolic capacity predicted by the genotype. Individualized dosing strategies may be characterized as the Cinderella of precision medicine – even if easily applied, they have rarely supplanted the one-dose-fits-all approach. Drug metabolism biomarker information for the purposes of dose adjustments is usually generalizable to all diseases where the same treatment is given and in principle generalizable to different patient or age groups. Note, however, that while the genotype remains stable, drug metabolism phenotypes may change over time, as when affected by drug–drug or drug–nutrients interactions, an effect referred to as phenocopying [Citation5]. In such cases, the amount of dose adjustment should take this effect into account [Citation6].
Many metabolism biomarkers also do double duty as companion diagnostics for both dose adjustments and safety. As an example, codeine (which is metabolized to morphine by the polymorphic enzyme CYP [cytochrome P450 enzyme] 2D6) is at high risk of causing breath depression in carriers of the CYP2D6 ultrarapid metabolizer genotype. This risk is especially acute in neonates, because in this age group, the limited glucuronidation capacity hampers the elimination of morphine [Citation7]. As the example demonstrates, care must be given to the interaction with other factors when these biomarkers are used for safety. Extraordinary high efavirenz concentrations in African patients led to the identification of genetic variants predicting low activity of CYP2B6 which is the main enzyme involved in metabolism of efavirenz. These low-activity alleles are highly prevalent in Africans but not in Caucasians [Citation8]. Dose adjustments therefore may differ across ethnicities and address safety issues related to high drug exposure [Citation9].
Drug response biomarkers establish the indication for therapy. These companion diagnostics are typically employed in connection with so-called ‘targeted therapies,’ particularly in cancer therapy. Often, testing prior to the initiation of therapy is mandatory as indicated in the drug labels. Most of these biomarkers are specific tumor genes, and therefore somatic mutations. A prototypical example is given by the Philadelphia chromosome translocation causing the bcr–abl fusion protein kinase in hematological malignancies, targeted by imatinib. Also germline biomarkers are represented in this group, such as the BRCA variants, mandatorily tested for the indication of poly-adenosine diphosphate (ADP) ribose polymerase (PARP) inhibitors.
The indication for therapy, as well as the indication for testing with an obligatory companion diagnostics, may refer to a specific disease and/or a stage in the disease progression (attention to extrapolation to other disease areas). In drug response companion diagnostics, generalizability across different patient groups differs from safety or drug metabolism markers, because administering the drug to patients other than those specified in the indication section of the drug label constitutes ‘off-label’ use of the drug, and possibly exposes the patient to unknown risks. Also the use of a drug in a different cancer type, or in a different disease stage, and even in a different therapy phase, carries an unknown risk/benefit ratio. Using a biomarker as companion diagnostics from one cancer type in a different tumor constitutes an off-label application of targeted drugs, in which the bioavailability of the drug at the target may not have been tested, potential interactions with the standard therapy may be unknown, and the significance of the biomarker for the oncogenetic process may differ. The validity of these companion diagnostics therefore may not be extrapolated to different cancer types, stages, or therapy phases.
Drug response monitoring or therapy surveillance biomarkers usually refer to clinical parameters such as cholesterol levels for statin therapy. They may also constitute surrogate end points in clinical studies. The new developments in precision medicine have added the genetic dimension to the field of surveillance biomarkers. Genetic surveillance biomarkers may include gene expression changes or epigenetic modifications in the target tissue [Citation10]. An example is given by liquid biopsy gene expression signatures in human platelets that may be used for companion diagnostics on tumor and disease state in individual patients [Citation10].
The trend toward accelerated drug approval that is taking hold in both the United States and Europe is facilitating the development of precision medicine drugs [Citation11]. In the United States, the regulatory framework offers the possibility of a combined market authorization of the companion diagnostics together with the drug with which it is intended to be used [3]. In this approach, the context of application of the companion diagnostic and the validity of the biomarker are evaluated as part of the regulatory process. In Europe, the in vitro diagnostics and in-house diagnostic tests are regulated separately from drugs in a specific framework for medicinal products [Citation12]. In both regulatory frameworks, the biomarker validation process includes systematically characterizing biomarker validity and clinical utility as prescribed in the guidelines for biomarker qualification in Europe or biomarker validation in the United States [Citation13,Citation14].
The availability of biomarkers at the time of marked access may seduce practitioners into an uncritical generalization of the information delivered by biomarkers outside their original application domain (dose range, safety risk, and drug effects in specific patient subgroups). As an example, genome-wide sequencing analysis of a tumor sample provides all the information on drug-sensitive targets in one test. Targeted drugs may be added to standard therapy without consideration for the possible uncertainties given by a knowledge base limited to the group or application domain in which the biomarker was originally investigated. If a standard therapy exists, the additional risks for safety and efficacy of the combined use of targeted therapies following biomarker testing may arise from unforeseen interactions. Further variability may arise, for example, when heterogeneous sources of material such as purified tumor tissue, tumor-surrounding tissue, biopsy material, blood, or plasma samples are used for the diagnostics. To overcome these barriers in common pharmacogenetic testing practice, we should focus on obtaining a well-established basis of evidence on analytic validation and clinical utility. Also, data on clinical biomarkers, comorbidities, and lifestyle habits that impact drug response should be collected. These data often are not considered at the stage of clinical trials for evaluation of clinical utility in biomarker tests. Diagnostic platforms with robust assay performance even in broad applications in general clinical laboratories may be preferable to promote the use of valid tests. Centralized testing facilities with standardized methods may promote the acquisition of valid data.
Precision medicine practice does not always consider issues of generalizability in biomarker testing as would be appropriate. To increase transparency in the intended use of companion diagnostics, it may be beneficial to consider formalizing the applicability of the output of biomarker tests in an accompanying description. Here, it should be specified for which drugs, dose range, disease or disease groups, and ethnicities the information delivered by the diagnostics has been validated. Similar to the issuing of the pharmacogenetic implementation consortium guidelines [Citation15], this companion document could be generated by extracting all available evidence for the use of the test and may aid communication of results of tests from diagnostic laboratories to clinicians in the care of individual patients. This would avert the danger to fall into a ‘one-test-fits-all’ approach.
Declaration of interest
The author has no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Additional information
Funding
References
- Relling MV, Evans WE. Pharmacogenomics in the clinic. Nature. 2015;526(7573):343–350.
- Eichler H-G, Abadie E, Breckenridge A, et al. Bridging the efficacy-effectiveness gap: a regulator’s perspective on addressing variability of drug response. Nat Rev Drug Discov. 2011;10(7):495–506.
- FDA. Guidance for industry: in vitro companion diagnostic devices. Silverspring (MD): FDA; 2014.
- PharmGKB. Internetquelle [cited 2016 May]. Available from: https://www.pharmgkb.org/view/drug-labels.do.
- Shah RR, Gaedigk A, Llerena A, et al. CYP450 genotype and pharmacogenetic association studies: a critical appraisal. Pharmacogenomics. 2016;17(3):259–275.
- Stingl JC, Brockmöller J, Viviani R. Genetic variability of drug-metabolizing enzymes: the dual impact on psychiatric therapy and regulation of brain function. Mol Psychiatry. 2013;18(3):273–287.
- Kirchheiner J, Schmidt H, Tzvetkov M, et al. Pharmacokinetics of codeine and its metabolite morphine in ultra-rapid metabolizers due to CYP2D6 duplication. Pharmacogenomics J. 2007;7(4):257–265.
- Wang J, Sönnerborg A, Rane A, et al. Identification of a novel specific CYP2B6 allele in Africans causing impaired metabolism of the HIV drug efavirenz. Pharmacogenet Genomics. 2006;16(3):191–198.
- Swart M, Skelton M, Ren Y, et al. High predictive value of CYP2B6 SNPs for steady-state plasma efavirenz levels in South African HIV/AIDS patients. Pharmacogenet Genomics. 2013;23(8):415–427.
- Best MG, Sol N, Kooi I, et al. RNA-seq of tumor-educated platelets enables blood-based pan-cancer, multiclass, and molecular pathway cancer diagnostics. Cancer Cell. 2015;28(5):666–676.
- Leyens L, Richer É, Melien Ø, et al. Available tools to facilitate early patient access to medicines in the EU and the USA: analysis of conditional approvals and the implications for personalized medicine. Public Health Genomics. 2015;18(5):249–259.
- EuropeanCommission. Directive 98/79/EC on IVD medical devices (IVDD) (10/1998). Bruxelles (Belgium): European Commisssion; 1998.
- FDA. Guidance for industry: biomarkers related to drug or biotechnology product development: context, structure, and format of qualification submissions. Silverspring (MD): FDA; 2011.
- EMA. Qualification of novel methodologies for drug development: guidance to applicants. London: EMA; 2014.
- Relling MV, Klein TE. CPIC: clinical pharmacogenetics implementation consortium of the pharmacogenomics research network. Clin Pharmacol Ther. 2011;89(3):464–467.