
Abstract
Inheritance of host resistance to blue butterfly (PBB1) and plume moth (PPM1) in interspecific mapping populations (F2, F3 and BC1) derived from a cross involving Cajanus cajan (cv. ICP-26) × Cajanus scarabaeoides (acc. ICPW-94) appeared to be under monogenic control either by a single major gene or a cluster of tightly linked genes. Bulked segregant analysis using 237 [85 simple sequence repeats (SSR), 143 random amplified polymorphic DNA (RAPD) and nine inter simple sequence repeats (ISSR)] parental polymorphic primers led to the identification of 43 markers that distinguished the resistant and susceptible bulks alike to parents, and which were also segregating among F2 progenies. Linkage analysis of these markers along with interaction phenotype score for both traits generated a linkage group consisting of 11 markers (two SSR, seven RAPD and two ISSR) and two trait loci (PBB1 and PPM1). This linkage group distributed over 133.9 cM with an average marker interval of 10.3 cM. The PBB1 and PPM1 loci were linked to each other by 11.2 cM (rf 0.110), and were flanked by ISSR marker UBC8722000 (15.9 cM), and RAPD marker OPA09910 (15.3 cM), respectively. On the basis of sequence homology of linked marker OPA09910 these two loci were assigned to chromosome 2 (CcLG02). Composite interval mapping led to the detection of two major quantitative trait loci (qPBB2.1 and qPPM2.1) controlling blue butterfly and plume moth resistance, respectively and the quantitative trait locus peaks coinciding with PBB1 and PPM1 loci on the map.
Introduction
Pigeonpea (Cajanus cajan (L.) Millsp., Fabaceae) is cultivated as one of the major grain legume crops on 4.92 million hectares of dry and tangential lands in semi-arid tropical and subtropical countries in south Asia, Africa, the Caribbean and Latin America. The global annual production is 3.65 million tons with an average yield of 898 kg/ha (FAOSTAT Citation2010). The annual yield of this crop has stagnated over the last couple of decades due to various biotic and abiotic stresses (Saxena Citation2008) together with unadapted agronomic practices and crop management (Varshney et al. Citation2012). Pigeonpea being a crop of warm humid semi-arid tropics, this crop became a suitable host for various oligo- and poly-phagous pests, mostly those sucking and feeding on inflorescences and pods. A group of such pests [Helicoverpa armigera Hubner (pod borer), Maruca testulalis Geyer (spotted pod borer), Lampides boeticus L. (blue butterfly), Exelastis atomosa Wals. (plume moth) and Melanogromyza obtuse Malloch. (pod fly)] constitutes a pod borer complex that poses major biotic constraints on achieving sustainable yield in pigeonpea (Grover and Pental Citation2003; Gnanesh et al. Citation2011). These pests cause 30–80% yield losses (Sharma et al. Citation2010), which accounts for 2.5 million tons of grains per annum, worth more than US$ 300 million per year (Banu, Muthiah, and Ashok Citation2005). Among these destructive pests, the major loss (70–80%) was due to H. armigera (CitationVerulkar, Singh, and Bhattacharya Citation1997; Banu, Muthiah, and Ashok Citation2005), and in addition by L. boeticus (4–10%) and E. atomosa (5–10%) (Sharma et al. Citation2010). The use of chemicals and pesticides is only partially effective in controlling these pests, and is ecologically harmful and financially not viable. The alternative approach to overcome this problem is the identification and subsequent introgression of host resistance into the promising cultivars, which is an efficient, speedy, reliable and safe means of crop protection. The sources of host resistance to the pod borer and its associated pests including plume moth and blue butterfly were not reported in the primary gene pool (cultivars and local landraces) of pigeonpea so far. However, some of the accessions of Cajanus scarabaeoides (L.) Thouars, which belongs to the secondary gene pool (wild, but crossable) possessed host resistance for several pests (Mallikarjuna, Saxena, and Jadhav Citation2011) including pod borer, caused by H. armigera (Verulkar, Singh, and Bhattacharya Citation1997; Aruna et al. Citation2005; Mishra et al. Citation2013). Furthermore, these authors reported single dominant genetic control for pod borer resistance in pigeonpea involving inter-specific segregating populations. Recently, a couple of DNA markers linked with pod borer resistance have also been identified in pigeonpea (Mishra et al. Citation2013).
The use of DNA markers in conventional plant breeding is useful for mapping, monitoring and cloning of agro-economic genes (Kole and Gupta Citation2004; Varshney, Graner, and Sorrells Citation2005), and the availability of a tightly linked marker for the desired trait paves the way for introgressive-marker-assisted breeding (Panigrahi et al. Citation2013). Identification of a DNA marker linked with blue butterfly and plume moth resistance would also allow prompt screening of cultivars, local land races, breeding lines and segregating populations before their use in breeding endeavours aimed at marker-assisted introgression of alleles. There is no report available either on their mode of inheritance or the genetic linkage of DNA markers with blue butterfly and plume moth resistance in pigeonpea.
This study reports on the identification of host resistance against blue butterfly and plume moth in genotypes of C. scarabaeoides, and their mode of inheritance using interspecific mapping populations, and identification of loci on a genetic linkage map based on simple sequence repeats (SSR), random amplified polymorphic DNA (RAPD) and inter simple sequence repeats (ISSR) markers.
Materials and methods
Screening of a collection of genotypes for host resistance to blue butterfly and plume moth
Seeds of 34 genotypes belonging to genus Cajanus, including seven cultivars of C. cajan and 27 genotypes of 11 wild species belonging to primary, secondary and tertiary gene pools of pigeonpea, were obtained from ICRISAT, Patancheru, Andhra Pradesh (Table ). These genotypes were maintained in the experimental garden of the School of Life Sciences, Sambalpur University, Odisha, India (21°29'5" N, 83°52'41" E; altitude 173 m above mean sea level).
Table 1. Host response of 34 genotypes of Cajanus spp. against blue butterfly (Lampides boeticus) and plume moth (Exelastis atomosa) in vivo (multi-choice field conditions).
In vivo screening of genotypes
Ten plants of each genotype were planted in a 2 × 2-m plot in the experimental garden during the third week of October 2010 so that the reproductive phase of the plants coincided with maximum population of pests during February–March 2011. For screening, two to three inflorescences per plant were tagged on the 15th day after flowering, and observations on percentage of buds, flowers and pods infected per inflorescence by either blue butterfly or plume moth were recorded in the field under multi-choice conditions at the end of the 30th day of tagging. For screening host resistance against blue butterfly and plume moth, the pods and inflorescence present on the tagged stem were screened for exit holes and pin holes, respectively. Accordingly, these genotypes were assigned with interaction phenotype (IP) score on a scale of 1–9 (Sahu et al. Citation2015), where 1 is completely resistant (no damage) and 9 is highly susceptible (≥ 35% pod damage). The genotypes with IP score 1 to 3 were considered resistant and those with IP score 4 to 9 were considered susceptible to both pests.
In vitro screening of genotypes
Larvae and adults of plume moth and blue butterfly were obtained from the colonies maintained in the laboratory for in vitro antibiosis assay. The colony of each pest was added separately with their natural population collected from the field to make the larvae populations heterogeneous. The larvae were reared on the chickpea-based artificial diet (Armes, Bond, and Cooter Citation1992) at 27 ± 1°C, 65 ± 5% relative humidity and 12-hour illumination using cool fluorescent light. The adults were released in 30 × 30 × 30-cm cages. Nappy liners hung inside the cages were provided as a substrate for oviposition and adults were fed with 10% sucrose solution in absorbent cotton. Eggs laid on the nappy liners were sterilized with 1% sodium hypochlorite solution, and transferred into 200-ml plastic cups smeared with a 2-mm-thick layer of artificial diet for rearing in groups of 100 to 150. After 5 days, the larvae were transferred to six-well plates (having 5 ml artificial diet in each well), and reared individually until pupation. The larvae were used for assessing antibiosis to plume moth and blue butterfly by incorporating lyophilized pod powder (10 g/l) into the artificial diet.
Expression of antibiosis was also assessed by rearing respective neonate larvae on artificial diet (Sujana, Sharma, and Rao Citation2008) augmented with lyophilized pod powder of 34 genotypes with few modifications (Mishra Citation2013). The blended artificial diet (c. 20 ml) was poured into small glass vials (50 × 100 mm) and fed to these larvae to assess antibiosis response. The rearing cups were kept at 27 ± 2°C, 65 ± 5% relative humidity and 12-hour photoperiod. Each treatment was replicated three times, and there were 10 larvae in each replication. Data on survival of larva and larval weights were recorded on the 10th day after initiating the experiment, as explained above. Similarly, data on weights of pupae were recorded after one day of pupation. Data were recorded on larval and pupal growth periods on the artificial diet. On the basis of larval growth 34 genotypes were assigned an in vitro IP score on the 1–9 scale, where 1, ≤ 1.0 mg increment in larval weight; 2, > 1.0 mg and ≤ 5.0 mg increment in larval weight; 3, > 5.1 mg and ≤ 10.0 mg increment in larval weight; 4, > 10.1 mg and ≤ 15.0 mg increment in larval weight; 5, > 15.1 mg and ≤ 20.0 mg increment in larval weight; 6, > 20.1 mg and ≤ 25.0 mg increment in larval weight; 7, > 25.1 mg and ≤ 30.0 mg increment in larval weight; 8, > 30.1 mg and ≤ 35.0 mg increment in larval weight; and 9, ≥ 35.1 mg and above increment in larval weight. Genotypes with score 1 to ≤ 3 were considered resistant, and the genotypes with score > 3 to 9 were considered susceptible.
Development of mapping populations for inheritance studies and linkage analysis
On the basis of plant–pest interaction in field conditions and expression of antibiosis, single plants of C. cajan cv. ICP-26 (in vivo IP score of 8 and 9 for plume moth and blue butterfly, respectively; in vitro IP score of 9 for both blue butterfly and plume moth; susceptible) and C. scarabaeoides acc. ICPW-94 (in vivo IP score of 1 for both plume moth and blue butterfly; in vitro IP score of 3 for both blue butterfly and plume moth; resistance), were self-pollinated and cross-hybridized under a net house during the post rainy season of 2011/12. A single F1 hybrid plant was self-pollinated to generate the F2 population (116 progenies), and was crossed with ICP-26 to generate 12 BC1 plants. F2 plants were individually self-pollinated to develop 78 F3 families.
Phenotyping of mapping populations for host resistance
Initially, 116 F2 progenies and 10 plants from each parent were evaluated for host resistance (field reaction and antibiosis assay) to plume moth and blue butterfly during the pre-rainy season of 2012/13. Twelve plants of the susceptible cultivar C. cajan cv. ICP11543 were planted as a control. During the pre-rainy season of 2013/14, the parents, BC1 plants and progenies of 78 F3 families were assessed for host resistance to both plume moth and blue butterfly in field conditions only. In addition, all the pods of individual plants were examined for the presence of both exit holes and pin holes at an interval of every 7 days. The evaluation entries were then scored on an IP scale of 1–9, as explained above.
Isolation and purification of genomic DNA
Genomic DNA was isolated from young leaves of parents and F2 progenies using modified cetyl-trimethyl-ammonium bromide (CTAB) protocol (Sivaramakrishnan et al. Citation1997), and were equilibrated to a final concentration of 10 ng/μl using T10E1 [10 mm Tris–HCl and 1 mm ethylene diaminetetraacetic acid (EDTA)] buffer. Equal amounts of DNA from ten highly resistant (IP score 1) and ten highly susceptible (IP score of 7 to 9) F2 progenies for plume moth and blue butterfly were pooled discretely to constitute the resistant bulk and susceptible bulk for both pests, respectively for bulked segregant analysis (Michelemore, Paran, and Kesseli Citation1991).
RAPD and ISSR marker analysis of the parents and mapping populations
For RAPD and ISSR analysis, polymerase chain reaction (PCR) amplification of 25 ng of genomic DNA was carried out separately using each of 150 decamer oligonucleotide primers (Operon Technologies, Alameda, CA, USA) and 12 synthesized ISSR primers from the set 100/9 (University of British Columbia, Vancouver, Canada), respectively. Each amplification reaction (25 μl) contained the 25-ng template DNA, 2.5 μl of 10× assay buffer [100 mm Tris–HCl pH 8.3; 0.5 m KCl; 0.1% (weight/volume; w/v) gelatine], 1.5 mm MgCl2, 200 μm of each dNTP, 0.25 μm primer, 1.0 units Taq DNA polymerase (Bangalore Genei Pvt. Ltd., India). Amplification conditions for RAPD analysis include an initial denaturation step of 94°C for 5 min, followed by 45 cycles of denaturation at 94°C for 1 min, primer annealing at 37°C for 1 min, and an extension at 72°C for 2 min; then a final extension was carried out at 72°C for 5 min (Williams et al. Citation1990). For ISSR analysis, the following PCR conditions were used – an initial denaturation step of 94°C for 5 min, followed by 40 cycles of denaturation at 94°C for 30 sec, a primer annealing at 40–60°C for 45 sec, and an extension at 72°C for 2 min; then a final extension was carried out at 72°C for 5 min (Mishra et al. Citation2013). Both RAPD and ISSR amplification was carried out in a thermal cycler (Master Cycler Pro; Eppendorf, Hamburg, Germany), and PCR products were separated in 1.4% (w/v) agarose gel containing 0.5 μg/ml −ethidium bromide in TAE buffer (40 mm Tris–acetate, pH 8.0; 2 mm EDTA) at a constant 50 V. A gel loading buffer [20% (w/v) sucrose, 0.1 m EDTA, 1.0% (w/v) sodium dodecyl sulphate, 0.25% (w/v) bromo-phenol blue, 0.25% (w/v) xylene cyanol] was used as a tracking dye. Amplified DNA fragments were visualized on the gel documentation system (Fire Reader Gel Doc System, Uvitec, Cambridge, UK) and photographed. The sizes of the amplified products were determined using a 250-bp step-up ladder (Bangalore Genei Pvt. Ltd., India) as standard, and TL-120 software (Non-linear Dynamics, Total Lab Ltd., Newcastle Upon Tyne, UK). Each marker locus was named by assigning the primer code followed by the size of the fragment in subscript.
SSR marker analysis of the parents and mapping populations
One hundred pairs of synthesized SSR primers (Dutta et al. Citation2011) were tested to identify polymorphism among ICP-26 and ICPW-94 for further genetic analysis. For SSR analysis, PCR amplifications were carried out in 15-μl volume containing 1.5 μl of 10× assay buffer [160 mm (NH4)2SO4, 500 mm Tris–HCl, pH 9.2; 17.5 mM MgCl2; 0.1% Triton X-100], 133 μm of dNTPs, 10 pmol each of forward and reverse primers, 50 ng of genomic DNA and 0.75 U of Taq DNA polymerase (Vivantis Technologies, Selangor, Malaysia). The PCR cycling profile was initial denaturation at 94°C for 5 min, followed by 35 cycles of 94°C for 1 min, 55°C for 1 min, 72°C for 1 min and a final extension at 72°C for 7 min. The PCR were performed using gradient PCR (Master Cycler Pro; Eppendorf, Hamburg, Germany). The amplified products were separated in 4% metaphor agarose gels (Lonza, Rockland, ME, USA) containing 0.1 μg/ml ethidium bromide in 1× TBE (40 mm Tris–borate, pH 8.0; 2 mm EDTA) buffer at 120 V for 2 h. A gel loading buffer [20% (w/v) sucrose; 0.1 m EDTA, 1.0% (w/v) sodium dodecyl sulphate; 0.25% (w/v) bromo-phenol blue; 0.25% (w/v) xylene cyanol] was used as a tracking dye. Amplified DNA fragments were visualized and molecular sizes of the amplified products were determined using a 100-bp step-up ladder (Bangalore Genei Pvt. Ltd.) as standard. Each marker locus was named by assigning the primer code (HASSR) followed by the number designated (Dutta et al. Citation2011).
Segregation and genetic linkage analysis using SSR, RAPD and ISSR markers
SSR (12), RAPD (26) and two ISSR primers distinguishing the parents (ICP-26 and ICPW-94) as well as the bulked samples (resistant bulk and susceptible bulk), were used for single plant analysis of F2 progenies. For the dominant markers, F2 individuals with ICP-26-specific amplified fragment were genotyped as ‘A’, and individuals without amplified fragments were genotyped as ‘B’. In contrast, F2 individuals with ICPW-94-specific amplified fragment were scored as ‘B’ and vice versa. For co-dominant markers, the F2 individuals with ICP-26-specific amplified fragment were genotyped as ‘A’, individuals with ICPW-94-specific amplified fragments were genotyped as ‘B’, and the genotype with both ICP-26- and ICPW-94-specific amplified fragments were genotyped as ‘H’. For phenotyping against plume moth and blue butterfly infestation, the F2 progenies were denoted as either ICP-26 type (A) or ICPW-94 type (B). For all cases the missing data were denoted as ‘–’ for genetic linkage analysis.
Linkage relationships among the segregating loci were established using the computer package MAPMAKER/EXP.3.0 (Lincoln, Dally, and Lander Citation1992). The linkage group was generated using all possible two-point recombination estimates with minimum LOD score of 4.0, and a recombination frequency of 0.30. The most probable marker order was determined using three-point tests followed by the ‘order’ command. The ‘ripple’ command was used to verify the marker order. Map distances were expressed in centiMorgan (cM) using the Kosambi map function (Kosambi Citation1943).
Quantitative trait locus analysis
Genotyping and phenotyping data against blue butterfly and plume moth resistance traits were analysed for mapping quantitative trait loci (QTLs) by using composite interval mapping (CIM; Zeng Citation1994) in the Windows QTL Cartographer, version 2.5 (Wang, Basten, and Zeng Citation2012). CIM analysis was performed using the standard model, scanning the genetic map and estimating the likelihood of a QTL and its corresponding effects at every 1 cM, while using significant marker cofactors to adjust the phenotypic effects associated with other positions in the genetic map. The number of marker cofactors for the background control was set by forward–backward stepwise regression. A window size of 5 cM was used, and therefore cofactors within 2.5 cM on either side of the QTL test site were not included in the QTL model. QTLs were determined to be significant if the corresponding likelihood ratio score was > 11.5 (equal to a LOD score of 2.5). The percent of phenotypic variance explained by a QTL was estimated at the highest probability peaks.
Cloning of linked DNA markers and sequence analysis
Flanking RAPD marker OPA09910 was excised from gels using a Genei Pure™ gel extraction kit (Bangalore Genei, Merck Bioscience, India) following the manufacturer’s instructions, and the eluted product was used for cloning and sequencing reactions. The purified DNA fragment was ligated into pTZ57R/T vector system and ligation product was transformed into Escherichia coli strain JM109 competent cells (Fermentas, Burlington, ON, Canada) using an InsTAclone™ PCR Cloning Kit (ThermoScientific, Waltham, MA, USA) and the manufacturer’s instructions. Cells were plated on pre-warmed Luria-Bertani (LB) agar medium containing ampicillin (50 μg/ml), 40 μl of X-gal (20 mg/ml) and 40 μl of isopropyl β-d-1-thiogalactopyranoside (IPTG) (100 mm) followed by overnight incubation at 37°C. The resulting white colonies on X-gal-IPTG-LB plates containing 50 μg/ml ampicillin were selected, being the putative recombinant clones.
The putative recombinant clones were grown in LB broth containing 50 μg/ml of ampicillin, and plasmids were isolated using a PureSol™ plasmid isolation kit (Bangalore Genei, Merck Bioscience). The plasmids were screened for their insert size by amplification using M13/pUC sequencing primers (S0100 and S0101) following the instructions depicted in the manual. The amplified products were electrophoresed in 1.4% agarose gel and visualized as explained earlier. The cloned fragments was sequenced using a dideoxy chain termination method using M13/pUC-S0100 and M13/pUC reverse S0101 primers at the DNA sequencing Facility (Eurofins Genomics India Pvt. Ltd., Bangalore, India) using a cycle sequencing method in ABI prism 3730 (Applied Biosystem, Foster City, CA, USA) on a hiring basis. The obtained sequence was submitted as query for Basic Local Alignment Search Tool (BLAST) analysis for homology with the deposited sequence of pigeonpea genome databases (http://www.ncbi.nlm.nih.gov/nuccore/ 450342093; http://www.ncbi.nlm.nih.gov/nuccore/ AFSP00000000.1).
Results
Screening of a collection of genotypes for host resistance to blue butterfly and plume moth
Screening under field conditions
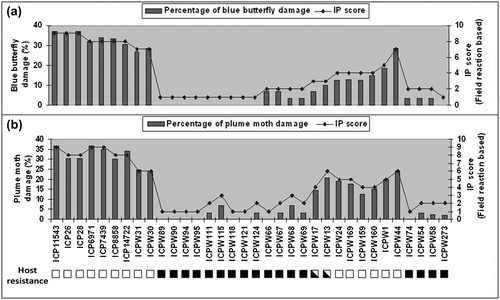
Among the 34 genotypes screened under field conditions, 15 wild genotypes belonging to four species of Cajanus [C. scarabaeoides, C. platycarpus (Benth.) van der Maesen, C. reticulatus (F.Muell.) van der Maesen and C. cinereus (F.Muell.) F.Muell.] showed consistent resistance against both pests with limited pod damage (IP score 1 to 3), whereas all seven genotypes of C. cajan appeared as conducive hosts for both pests, and so were grouped under ‘susceptible’ with IP scores of 8 and 9 (Table and Figure ). The rest of the genotypes of the secondary and tertiary gene pool showed different degrees of resistance and/or susceptibility to these pests with an IP score varying from 3 to 7.
Figure 1. Graphical sketch showing field resistance to blue butterfly (a) and plume moth (b) in terms of percentage of pod damage vis-a-vis IP score of 34 genotypes of Cajanus spp. (■ resistant to both pests, □, susceptible to both pests and ⬕, susceptible to one of the pests).
Screening through in vitro antibiosis assay
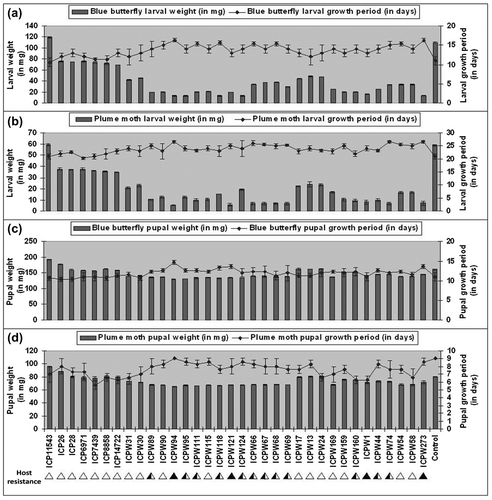
There were significant differences in survival rate, larval weight and pupal weight of blue butterfly and plume moth grown on an artificial diet augmented with pod powders of different genotypes inter se. The survival rate of plume moth and blue butterfly larvae on an artificial diet based on pod powders of seven cultivars of C. cajan varied from 83.3 ± 0.57% to 100.0%, and in the case of the 27 genotypes of the 11 wild species, it varied from 19.6 ± 0.48% to 65.6 ± 0.57% and 20.3 ± 0.57% to 53.3 ± 0.57%, respectively. The average weight of larvae and pupae and the growth period of the two different larvae on artificial diet are enumerated in Table and Figure . The plume moth larvae took 23–27 days to complete their development when reared on artificial diet supplemented with pod powders of wild genotypes, compared with 19–23 days with cultivars. The pupal period lasted for 8–9 days for most of the genotypes of C. scarabaeoides (including ICPW-94) when reared on artificial diets with pod powders similarly to other genotypes (Figure d). A similar trend was also observed for the growth of larvae and pupae of blue butterfly (Table and Figure ). On the basis of survival and growth of larvae the genotypes were assigned with an IP score on a 1 to 9 scale (Table ).
Table 2. Survival, growth and development of blue butterfly (Lampides boeticus) and plume moth (Exelastis atomosa) larvae and pupae on artificial diet with lyophilized pod powder of 34 genotypes of Cajanus species used in this study.
Figure 2. Graphical sketch showing the differences in larval weight (on 10th day), larval growth period, pupal weight and pupal growth period of blue butterfly (a, c) and plume moth (b, d) on AD supplemented with pod powders of 34 genotypes of Cajanus spp.
Inheritance of host resistance to blue butterfly and plume moth
Following in vivo and in vitro screening, two genotypes C. cajan cv. ICP-26 and C. scarabaeoides acc. ICPW-94 were chosen to raise the mapping population. All four F1 plants generated from the cross (C. cajan cv. ICP-26 × C. scarabaeoides acc. ICPW-94) were resistant to plume moth and blue butterfly with an IP score ‘1’. On the basis of simple phenotyping as ‘resistant’ and ‘susceptible’, frequency distribution of 116 F2 progenies gave a good fit to 3 : 1 for blue butterfly resistance (χ2 = 2.253; p = 0.133) and plume moth resistance (χ2 = 1.655; p = 0.198), indicating single dominant gene control for both blue butterfly and plume moth resistance (Figure and Table ). In the F3 generation, the segregation pattern of 78 F3 families for blue butterfly reaction was a good fit for 1 (20 non-segregating resistant) : 2 (42 segregating) : 1 (16 non-segregating susceptible) ratio (χ2 = 0.872, p = 0.832), which was also evidence for oligo-genic control of blue butterfly resistance. Similar to the segregation seen for blue butterfly, 78 F3 progenies were also segregated in a 1 (18 non segregating resistant) : 2 (46 segregating) : 1 (14 non-segregating susceptible) ratio (χ2 = 2.923, p = 0.403) for plume moth resistance, showing a monogenic segregation pattern for plume moth resistance. Again a 1 : 1 segregation pattern at BC1 for both blue butterfly (χ2 = 1.334, p = 0.248) and plume moth (χ2 = 0.332, p = 0.564) resistance confirmed their single dominant gene action. The joint segregation of host resistance to blue butterfly and plume moth also showed significant deviation from the independent assortment ratio (χ2 = 26.037, p ≤ 0.0001) and were positively associated (Table ). This was also evidenced by both association analysis (χ2 of independence values 24.64 with LOD 8.66) and correlation statistics (r2 = 0.89).
Figure 3. Frequency distribution F2 lines derived from cross involving Cajanus cajan (ICP-26) and Cajanus scarabaeoides (ICPW-94) for IP score with field resistance to blue butterfly and plume moth.
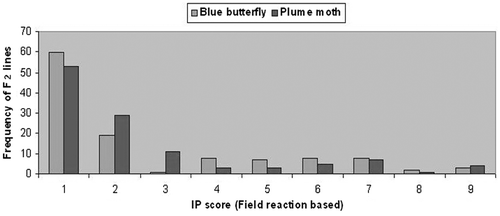
Table 3. Segregation* and linkage analysis of an interspecific F2 mapping population, involving Cajanus cajan (cv. ICP-26) and Cajanus scarabaeoides (acc. ICPW-94), for resistance to plume moth and blue butterfly.
Genetic linkage mapping of loci conferring resistance to blue butterfly and plume moth
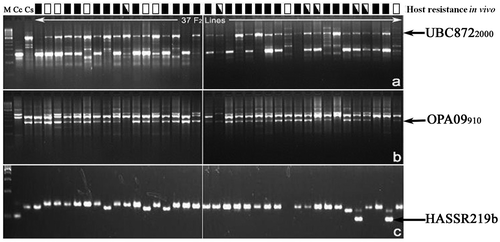
Among the 262 primers tested, 237 (85 pairs of SSR, 143 RAPD and nine ISSR) primers distinguished the parental lines and were applied for bulked segregant analysis. Out of them, only 40 parental polymorphic primers, including 12 SSR, 26 RAPD and two ISSR primers, differentiated resistant bulk and susceptible bulk as in the parent (Figure and Table ), and generated 43 DNA markers (10 SSR, 30 RAPD and three ISSR markers). Of these, 24 markers displayed a simultaneous bulk level polymorphism for both of the pests. Twenty markers including five SSR, 13 RAPD and two ISSR markers were linked to blue butterfly resistance in coupling phase, whereas nine RAPD, one ISSR and five SSR markers were in repulsion phase. Similarly, 16 markers including five SSR, nine RAPD and two ISSR markers were also linked to plume moth resistance in coupling phase, whereas 10 RAPD, one ISSR and five SSR markers were in repulsion phase. Among these markers, 15 (seven SSR, six RAPD and two ISSR) were distorted in their segregation (Table ). The segregation patterns of three markers, HASSR219, UBC8722000 and OPA09910, in a set of 37 F2 progenies together with host resistance to plume moth and blue butterfly are represented in Figure .
Figure 4. Bulked segregant analysis of F2 lines using random amplified polymorphic DNA primers – OPA19 and OPE09 (a), ISSR primers – UBC873 and UBC872 (b) and SSR primers HASSR 300 and HASSR 301 (c). Lanes: Cs = resistant parent (Cajanus scarabaeoides, acc. ICPW-94), RB = resistant bulk, Cc = susceptible parent (Cajanus cajan cv. ICP-26), SB, susceptible bulk; M, 250 bp (a,b) and 100 bp (c) step up ladder, respectively.
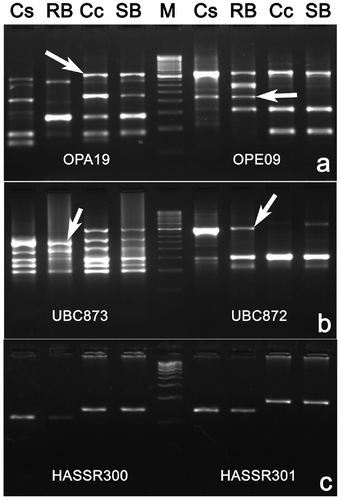
Table 4. Information about DNA markers generated by 40 polymorphic primers [RAPD (26) and ISSR (02) and SSR (12)] and their validation through bulked segregant analysis before single plant analysis for linkage studies.
Figure 5. Segregation of ISSR marker UBC8722000 (a), random amplified polymorphic DNA marker OPA09910 (b) and SSR marker HASSR219b (c) in a set of 37 F2 progenies. Lanes: M, 250 bp step up ladder, Cc = susceptible parent (Cajanus cajan cv. ICP-26) and Cs = resistant parent (Cajanus scarabaeoides, acc. ICPW-94). Reaction to blue butterfly and plume moth (■, resistant to both pests, □, susceptible to both pests and, susceptible to one of the pests) was indicated on the top of the lanes of 37 F2 lines.
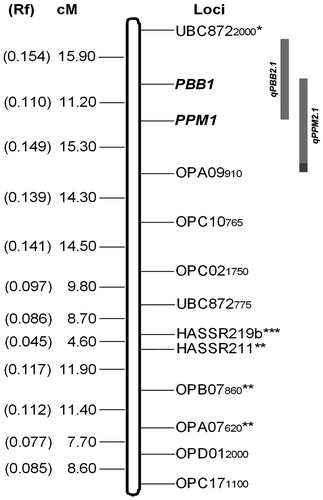
Linkage analysis at LOD of 4.0 and recombination frequency 0.3 grouped 11 of the 43 markers identified in the present study in one linkage group along with the loci conferring resistance to blue butterfly (PBB1) and plume moth (PPM1). This linkage group spanned over 133.9 cM with average marker interval of 10.3 cM. Five markers, including two SSR (HASSR211, HASSR219b), two RAPD (OPB07860, OPA07620) and one ISSR (UBC8722000), showed that segregation distortion remained in this linkage group (Table ). The PBB1 and PPM1 loci were loosely linked (11.2 cM), and flanked by UBC8722000 and OPA09910 at 15.9 cM and 15.3 cM, respectively (Figure ). Both the markers UBC8722000 and OPA09910 were linked to blue butterfly and plume moth resistance in coupling phase with recombination frequency of 0.154 (15.9 cM) and 0.149 (15.3 cM), respectively. However, double recombinants (13.79%) between UBC8722000 and OPA09910 loci were observed in lower frequency in this F2 population in comparison to single crossovers between PBB1 and PPM1 (being linked with recombination frequency 0.110) or with either of the flanked markers. For identification of chromosomal location, RAPD marker OPA09910 was cloned and sequenced. Sequencing of the marker OPA09910 revealed its length to be 942 bp with 41.3% GC content (GenBank accession no. KJ806291.1). BLAST analysis of this sequence with pigeonpea genome sequence showed its position on CcLG02, Scafseq.LG_V5.0fa: 25623599–25622679 bp (Varshney et al. Citation2012) and contig 01597, AFSP01.fsa.1: 8557–7640 bp (Singh et al. Citation2012) with more than 99% homology for 2–922 bp sequence of the marker. Hence, the probable chromosome number CcLG02 was assigned to this linkage group.
Table 5. Segregation of seven RAPD, two ISSR and two SSR markers (present in the linkage group) in the F2 interspecific mapping population along with genetic phase of linkage.
Figure 6. Linkage group indicating the position of DNA markers linked to the locus conferring resistance (PBB1) and plume moth (PPM1) in pigeonpea along with the their respective quantitative trait loci (qPBB2.1 and qPPM2.1); * deviated significantly from 3 : 1 segregation ratio at p = 0.5.
This framework map was used for composite interval mapping to depict the genetic contribution of the genomic locations containing resistant loci for blue butterfly or plume moth or both. Scanning the framework linkage map of 13 markers identified one major QTL (qPBB2.1) between the confidence interval UBC8722000 + 3.3 cM and OPA09910 + 15.4 cM at LOD score 41.3 for blue butterfly resistance. This locus explained 74.51% of phenotypic variation with an additive effect –11.801. Similarly, another QTL (qPPM2.1) was identified between the confidence interval UBC8722000 + 15.6 cM and OPA09910 + 0.1 cM at LOD score 45.17 for plume moth resistance. This QTL also explained 69.82% of phenotypic variation with additive effect –12.344. Both the QTL peaks coincided with the major loci identified by a simple phenotyping based linkage analysis (Figure ).
Discussion
Screening of host resistance for blue butterfly and plume moth, and their gene action
Blue butterfly and plume moth infestation inflated the loss in yield caused by pod borer in pigeonpea. Host plant resistance could be considered the preferred way of managing these pests, and so extensive efforts were made in the last couple of decades to identify the sources of host resistance among the germplasm of pigeonpea. On screening of host resistance among 34 promising genotypes, C. scarabaeoides acc. ICPW-94 showed consistent resistance against plume moth and blue butterfly in field conditions in vivo, and was also congruous with the antibiosis expression in vitro. Moreover, several accessions of C. scarabaeoides have already been identified and used as a source of host resistance to pod borer, pod fly and pod wasp in earlier studies (Verulkar, Singh, and Bhattacharya Citation1997; Sharma, Pampapathy, and Reddy Citation2003; Aruna et al. Citation2005; Sujana, Sharma and Rao Citation2008; Mishra et al. Citation2013; Rath et al. Citation2015).
Knowledge of inheritance and number of genes governing the host resistance to blue butterfly and plume moth is essential to formulate efficient breeding strategies in pigeonpea. Hence, the F1 plants, F2 interspecific progenies and BC1 lines, derived from a cross involving C. cajan cv. ICP-26 (susceptible) and C. scarabaeoides acc. ICPW-94 (Resistant), were used in the present attempt. All four F1 plants were resistant to both plume moth and blue butterfly infestation with an IP score of ‘1’ and their segregation among 116 F2 progenies gave a good fit to an expected 3 : 1 ratio, which were indicative of a single dominant gene control for both blue butterfly and plume moth resistance. The segregation patterns of 78 F3 families (1 : 2 : 1) and 12 BC1 plants (1 : 1) were also substantiated for monogenic control of host plant resistance against both blue butterfly and plume moth, as reported for pod borer resistance (Verulkar, Singh, and Bhattacharya Citation1997; Aruna et al. Citation2005; Mishra et al. Citation2013) and fusarium wilt resistance (Kotresh et al. Citation2006) in pigeonpea. However, the frequency distribution in F2 progenies with IP score 1 to 9 could not exclude the possible involvement of one or more genes for the host resistance conferred by C. scarabaeoides acc. ICPW-94. This might be due to simple plant–pest interaction phenotyping such as scoring ‘1’ and ‘0’ for resistant and susceptible in F2 population, used in this study and this approach will tend to estimate only the major gene(s) governing host resistance. Moreover, the joint segregation studies among host resistance conferred by these two pests deviated significantly from expected 9 : 3 : 3 : 1, which was also an indication towards close linkage between the two loci contributing host resistance and was further confirmed by association analysis.
Molecular mapping of loci conferring blue butterfly and plume moth resistance
The application of bulked segregant analysis with DNA markers including SSR, ISSR and RAPD can facilitate the detection of markers closely linked to the trait of agronomic interest (Michelemore, Paran, and Kesseli Citation1991). The segregation of host plant resistance in F2 progenies and dominant RAPD and ISSR markers in the coupling phase of linkage showed good fit to 3 : 1 ratio baring two RAPD markers (OPB07860 and OPA07620) and one ISSR marker (UBC8722000). The segregation distortion of a few dominant markers could also be attributed to abortion of male or female gametes or selective exclusion of particular gametic genotypes from fertilization, owing to incompatibility, incongruity and preferential zygote selection during interspecific hybridization involving C. cajan cv. ICP-26 and C. scarabaeoides acc. ICPW-94 (Kreike and Stiekema Citation1997; Gupta et al. Citation2006). The codominant SSR marker could distinguish homozygous and heterozygous resistant genotypes among the segregating progenies (Zietkiewicz, Rafalski, and Labuda Citation1994). In our study, the difference among homozygous and heterozygous resistant plants was depicted by both the SSR markers, except for some F2 progenies, which points towards the involvement of a common genomic region conferring resistance to both blue butterfly and plume moth. Moreover, these findings also showed putative genetic linkage of these markers with blue butterfly and plume moth resistance.
Marker-assisted breeding is the foremost application of DNA markers, and one of its primary prerequisites is the establishment of marker to gene linkage through segregating populations (Panigrahi et al. Citation2013). Identification of DNA marker(s) closely linked to blue butterfly and plume moth resistance should facilitate future breeding efforts aimed at introgression of corresponding alleles in pigeonpea. Only a few reports pertaining to identification of DNA markers linked to qualitative traits, namely fusarium wilt resistance (Kotresh et al. Citation2006), sterility mosaic disease resistance (Ganapathy et al. Citation2009), plant type (Dhanasekar, Dhumal, and Reddy Citation2010) and pod borer resistance (Mishra et al. Citation2013) were available in pigeonpea. In the present study two loci, PBB1 and PPM1, were loosely linked (11.2 cM), and were flanked by two markers UBC8722000 and OPA09910 with the lowest number of recombinants between themselves. These findings supported the simultaneous use of both UBC8722000 and OPA09910 in marker-assisted breeding efforts using C. scarabaeoides acc. ICPW-94 as donor parent. Besides, the closely linked markers could be converted to sequence characterized amplified region (SCAR) markers for efficient and reliable introgression of host resistance alleles into the cultivated genetic background without antibiosis expression assay and field resistance screening. As both the markers were linked to PBB1 and PPM1 loci at more than 10 cM, only the sequence information of OPA09910 was deduced for chromosome assignment. This may further be used to search closely linked markers using genome sequence data of pigeonpea. On the basis of OPA09910 sequence homology with whole genome sequence (http://www.ncbi.nlm. nih.gov/nuccore/450342093) of C. cajan, both loci (PBB1 and PPM1) were assigned to CcLG02 (Varshney et al. Citation2012). This type of comparative genome analysis was first used for mapping of blast resistance gene in rice (Sharma et al. Citation2005, Citation2012) and black rot resistance locus in Brassica oleracea var. botrytis L. (Saha et al. Citation2014). Moreover, the scanning of this CcLG02 genome sequence may lead to the development of markers that are closely linked to the host resistance loci, which could help the introgression of alleles with more precision. CIM analyses also identified two major QTLs (qPBB2.1 and qPPM2.1) controlling blue butterfly and plume moth resistance, and these QTLs inherited the ‘resistant’ allele from the donor parent C. scarabaeoides acc. ICPW-94. Both the loci (qPBB2.1 and qPPM2.1) explained 74.51% and 69.82% variation at LOD values of 41.3 and 45.17, respectively. This is not uncommon and has been reported in many crop species including pigeonpea (Gnanesh et al. Citation2011; Kumawat et al. Citation2012). Further studies on fine mapping, positional cloning of these loci contributing host resistance against plume moth and blue butterfly, and their epigenome studies elucidate the molecular mechanism of host resistance in pigeonpea.
Conclusion
This study is the first report on the mode of inheritance of host resistance against blue butterfly and plume moth in pigeonpea, and these resistances appeared to be under the control of a major gene, but the frequency distribution pattern vis-à-vis the QTL analysis suggested the involvement of several minor genes. Both the major resistance gene loci (PBB1 and PPM1) along with 11 DNA markers constitute a linkage group, and the linkage group spans over 133.9 cM with an average marker interval of 10.3 cM. The PBB1 and PPM1 loci were linked to each other by 11.2 cM (rf 0.110), and were flanked by UBC8722000 (15.9 cM), and OPA09910 (15.3 cM), respectively. On the basis of sequence homology of the linked marker OPA09910, these two resistance loci were assigned to chromosome 2 (CcLG02).
Notes on contributors
Ramya Ranjan Mishra was born in Bhanjanagar, Ganjam, Odisha, India, in 1983. He received an MSc degree in Biotechnology from the Berhampur University, India in 2005 and a PhD in 2014 from Utkal University, India. Contribution: made crosses, generated the mapping population, genotyped and phenotyped the mapping population using SSR, RAPD and ISSR markers, and cloned the linked marker for sequencing. Read and approved the final manuscript.
Alok Ranjan Sahu was born in Sohela, Bargarh, Odisha, India in 1986. He received an MSc degree in Biotechnology and M.Phill degree in Life Sciences from Sambalpur University, India, in 2009 and 2010, respectively. Recently, Mr Sahu submitted his PhD dissertation to the Sambalpur University during July 2015. Contribution: made crosses, generated the mapping population, genotyped and phenotyped the mapping population using SSR, RAPD and ISSR markers. Interpreted the data and prepared the manuscript along with JP. Read and approved the final manuscript.
Sobha Chandra Rath was born in Chhandapali, Attabira, Bargarh, Odisha, India in 1988. He received an MSc degree in Biotechnology from Sambalpur University, India, in 2010. He is currently pursuing his PhD at Sambalpur University. Contribution: made crosses, generated the mapping population, genotyped and phenotyped the mapping population using SSR, RAPD and ISSR markers. Read and approved the final manuscript.
Jogeswar Panigrahi was born in Binika, Bolangir, Odisha, India in 1975. He received his MSc and PhD degrees from Utkal University in 1996 and 2005, respectively. He is recipient of KSKRA Award from BRNS, DAE Govt. of India. He has guided four PhD and more than 15 MPhil Scholars. He also successfully completed six R&D projects sponsored by different funding agencies. Dr Panigrahi is currently working on the plant molecular genetics of pigeonpea and in vitro conservation of medicinal plants. Contribution: conceived the project; Cloned the linked marker for sequencing. Interpreted the data and prepared the manuscript along with ARS. Read and approved the final manuscript.
Disclosure statement
There is no potential conflict of interest.
Funding
This work was supported by the Department of Science and Technology, Govt. of India [SR/FT/LS-92/2009] and Department of Biotechnology, Govt. of India[BT/PR13468/AGR/02 /702/2010]
Acknowledgements
The authors are grateful to the Department of Science and Technology, and the Department of Biotechnology, Govt. of India, New Delhi for financial assistance to the corresponding author (JP) though R&D projects (No. SR/FT/LS-92/2009 of DST and F. No. BT/PR13468/AGR/02 /702/2010 of DBT). The authors also gratefully acknowledge Dr H.D. Upadhyaya, Principal Scientist, ICRISAT for the germplasm and Vice Chancellor, Sambalpur University for providing necessary facilities.
References
- Armes, N. J., G. S. Bond, and R. J. Cooter. 1992. “The laboratory culture and development of Helicoverpa armigera.” Natural Resources Institute, Chatahm, China, bulletin no. 57: 1–21.
- Aruna, R., M. Rao, L. J. Reddy, H. D. Upadhyaya, and H. C. Sharma. 2005. “Inheritance of trichomes and resistance to pod borer (Helicoverpa armigera) and their association in interspecific crosses between cultivated pigeonpea (Cajanus cajan) and its wild relative, C. scarabaeoides.” Euphytica 145: 247–257.
- Banu, M. R., A. R. Muthiah, and S. Ashok. 2005. “Evaluation of pigeonpea (Cajanus cajan L.) genotypes against gram pod borer (Helicoverpa armigera)”, in Proceedings of 4th International Food legume Research Conference on Food legumes for Nutritional Security, ed. M. C. Kharkwal, 317–320. New Delhi: The Indian Society of Genetics and Plant Breeding.
- Dhanasekar, P., K. N. Dhumal, and K. S. Reddy. 2010. “Identification of RAPD markers linked to plant type gene in pigeonpea.” Indian Journal of Biotechnology 9: 58–63.
- Dutta, S., G. Kumawat, B. P. Singh, D. K. Gupta, S. Singh, V. Dogra, K. Gaikwad, et al. 2011. “Development of genic-SSR markers by deep transcriptome sequencing in pigeonpea (Cajanus cajan (L.) Millspaugh).” BMC Plant Biology 11: 17.
- FAOSTAT. 2010. (Food and Agriculture Organisation Statistics), FAO Uganda information bulletin. http://faostat.fao.org/faostat (accessed 11 February 2013).
- Ganapathy, K. N., G. M. Byre, S. C. Venkatesha, R. Ramachandra, B. N. Gnanesh, and G. Girish. 2009. “Identification of AFLP markers linked sterility mosaic disease in pigeonpea Cajanus cajan (L.) Millsp.” International Journal of Integrative Biology 7: 145–149.
- Gnanesh, B. N., A. Bohra, M. Sharma, M. Byregowda, S. Pandey, V. Wesley, R. K. Saxena, et al. 2011. “Genetic mapping and quantitative trait locus analysis of resistance to sterility mosaic disease in pigeonpea [Cajanus cajan (L.) Millsp.].” Field Crops Research 123: 53–61.
- Grover, A., and D. Pental. 2003. “Breeding objectives and requirements for producing transgenics for major field crops of India.” Current Science 84: 310–320.
- Gupta, S. K., A. Charpe, S. Koul, Q. M. R. Haque, and K. V. Prabhu. 2006. “Development and validation of SCAR markers co-segregating with an Agropyron elongatum derived leaf rust resistance gene Lr24 in wheat.” Euphytica 150: 233–240.
- Kole, C., and P. K. Gupta. 2004. “Genome mapping and map based cloning.” In Plant breeding: Mendelian to molecular approaches, edited by H. K. Jain and M. C. Kharkwal, 257–299. New Delhi: Narosa Publishing House.
- Kosambi, D. 1943. “The estimation of map distance from recombination values.” Annual Eugenics 12: 172–175.
- Kotresh, H., B. Fakrudin, S. M. Punnuri, B. K. Rajkumar, M. Thudi, H. Paramesh, H. Lohithaswa, and M. S. Kuruvinashetti. 2006. “Identification of two RAPD markers genetically linked to a recessive allele of a Fusarium wilt resistance gene in pigeonpea (Cajanus cajan L. Millsp.).” Euphytica 149: 113–120.
- Kreike, C. M., and W. J. Stiekema. 1997. “Reduced recombination and distorted segregation in Solanum tuberosum (2x) x S. spegazzinii (2x) hybrid.” Genome 40: 180–187.
- Kumawat, G., R. S. Raje, S. Bhutani, J. K. Pal, A. S. V. C. R. Mithra, K. Gaikward, T. R. Sharma, and N. K. Singh. 2012. “Molecular mapping of QTLs for plant type and earliness traits in pigeonpea (Cajanus cajan L. Millsp.).” BMC Genetics 13: 84.
- Lincoln, S. E., M. J. Dally, E. S. Lander. 1992. Constructing genetic linkage maps with MAPMAKER/EXP-3.0, Whitehead Institute Technical Reports (3rd ed.), Cambridge, MA, USA: Bet Distribution 3B.
- Mallikarjuna, N., K. B. Saxena, and D. R. Jadhav. 2011. “Cajanus.” In Wild Crop Relatives: Genomic and breeding resources Legume Crops and Forages, edited by C. Kole, 21–34. Dusseldorf, Germany: Springer Verlag.
- Michelemore, R. W., I. Paran, and R. V. Kesseli. 1991. “Identification of markers linked to disease resistance genes by bulked segregant analysis: A rapid method to detect markers in specific genomic regions by using segregating populations.” Proceedings of the National Academy of Sciences of the USA 88: 9828–9832.
- Mishra, R. R. 2013. “Molecular mapping of gene(s) conferring resistance to pod borer and pod fly in Cajanus cajan (L.) Millsp.” Ph.D Thesis, Utkal University, Bhubaneswar, India.
- Mishra, R. R., A. R. Sahu, S. C. Rath, B. Behera, and J. Panigrahi. 2013. “Molecular mapping of locus controlling resistance to Helicoverpa armigera (Hubner) in Cajanus cajan L. (Millspaugh) using interspecific F2 mapping population.” Nucleus 56: 91–97.
- Panigrahi, J., R. R. Mishra, A. R. Sahu, S. C. Rath, and C. Kole. 2013. “Marker-assisted breeding for stress resistance in crop plants.” In Molecular Stress Physiology of Plants, edited by G. R. Rout and A. B. Das, 387–426. New Delhi, India: Springer.
- Rath, S. C., A. Mishra, R. R. Mishra, A. R. Sahu, and J. Panigrahi. 2015. “Inheritance and mapping of protein markers detecting polymorphism among Cajanus cajan and C. scarabaeoides genotypes with contrasting host response to pod borer (Helicoverpa armigera).” Caryologia 68: 217–224.
- Saha, P., P. Kalia, H. Sonah, and T. R. Sharma. 2014. “Molecular mapping of black rot resistance locus Xca1bo on chromosome 3 in Indian cauliflower (Brassica oleracea var. botrytis L.).” Plant Breeding 133: 268–274.
- Sahu, A. R., R. R. Mishra, S. C. Rath, and J. Panigrahi. 2015. “Construction of interspecific genetic linkage map of pigeonpea using SCoT, RAPD, ISSR markers and simple inherited trait loci.” The Nucleus 58: 23–31.
- Saxena, K. B. 2008. “Genetic improvement of pigeonpea – a review.” Tropical Plant Biology 1: 159–178.
- Sharma, H. C., G. Pampapathy, and L. J. Reddy. 2003. “Wild relatives of pigeonpea as a source of resistance to the pod fly (Melanagromyza obtuse Malloch) and pod wasp (Tanaostigmodes cajaninae La Salle).” Genetic Resources Crop Evolution 50: 817–824.
- Sharma, O. P., J. B. Gopali, S. Yelshetty, O. M. Bambawale, D. K. Garg, and B. B. Bhosle. 2010. Pests of pigeonpea and their management. LBS building, IARI, New Delhi, India: NCIPM.
- Sharma, T. R., A. K. Rai, S. K. Gupta, J. Vijayan, B. N. Devanna, and S. Ray. 2012. “Rice blast management through host–plant resistance: retrospect and prospects.” Agricultural Research 1: 37–52.
- Sharma, T. R., M. S. Madhav, B. K. Singh, P. Shanker, T. K. Jana, V. Dalal, A. Pandit, et al. 2005. “High-resolution mapping, cloning, and molecular characterization of the Pi-kh gene of rice, which confers resistance to Magnaporthe grisea.” Molecular Genetics and Genomics 274: 569–578.
- Singh, N. K., D. K. Gupta, K. J. Pawan, K. M. Ajay, S. Dutta, S. Singh, S. Bhutani, et al. 2012. “The first draft of the pigeonpea genome sequence.” Journal of Plant Biochemistry and Biotechnology 21: 98–112.
- Sivaramakrishnan, S., K. Seetha, A. Nageshwar Rao, and L. Singh. 1997. “RFLP analysis of cytoplasmic male-sterile lines of pigeonpea (Cajanus cajan (L.) Millsp.) developed by interspecific crosses.” Euphytica 93: 307–312.
- Sujana, G., H. C. Sharma, and D. Manohar Rao. 2008. “Antixenosis and antibiosis components of resistance to pod borer, Helicoverpa armigera in wild relatives of pigeonpea.” International Journal of Tropical Insect Science 28: 191–200.
- Varshney, R. K., A. Graner, and M. E. Sorrells. 2005. “Genomics assisted breeding for crop improvement.” Trends Plant Science 10: 621–630.
- Varshney, R. K., W. Chen, Y. Li, A. K. Bharti, R. K. Saxena, J. A. Schlueter, T. A. Mark, et al. 2012. “Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers.” Nature Biotechnology 30: 83–89.
- Verulkar, S. B., D. P. Singh, and A. K. Bhattacharya. 1997. “Inheritance of resistance to pod fly and pod borer in the interspecific cross of pigeonpea.” Theoretical and Applied Genetics 95: 506–508.
- Wang, S., C. J. Basten, and Z. B. Zeng. 2012. Windows QTL Cartographer 2.5. Raleigh, NC: Department of Statistics, North Carolina State University.
- Williams, J. G. K., A. R. Kubelik, K. J. Livak, J. A. Rafalaski, and S. V. Tingey. 1990. “DNA polymorphism amplified by arbitrary primers are useful as genetic markers.” Nucleic Acids Research 13: 6531–6533.
- Zeng, Z. B. 1994. “Precision mapping of quantitative trait loci.” Genetics. 136: 1457–1468.
- Zietkiewicz, E., A. Rafalski, and D. Labuda. 1994. “Genome fingerprinting by simple sequence repeat (SSR) – anchored polymerase chain reaction amplification.” Genomics 20: 176–183.