
Abstract
Global herbarium collections house a vast number of plant specimens stretching back over centuries. They include rare and extinct species, important historical collections, and valuable type specimens that could help to resolve long-standing taxonomic issues. Historical plant collections also provide unique windows into past evolutionary processes and population histories that do not exist with modern data alone. However, because the DNA in such specimens is often degraded and scarce, manipulation and analysis of their genetic material can be challenging. Recent developments in non-destructive genetic sampling and working with very small quantities of genomic DNA, especially in next-generation sequencing and bioinformatic analysis of ancient DNA, now make the majority of herbarium specimens potentially accessible to phylogenetic, population genetic, and barcoding studies. For example, studies of DNA from long time series of Ipomoea batatas and Anacamptis palustris collections revealed linkages between anthropogenic activity and changes in haplotype distribution. These time series also enable the identification of genes under recent selection in the genomes of both the plants and their pathogens, as was shown for Phytophthora infestans, a microbial pathogen of Solanum tuberosum. Here, we summarize the major challenges in using historical plant DNA in evolutionary studies and review genetic studies integrating herbarium specimens. We expect future genetic studies of historical herbarium specimens to use genomic, metagenomic, and population genetic approaches to: investigate how plant populations respond to environmental change; infer temporal changes in genetic diversity; identify genes under recent selection; and investigate past plant pathogen epidemics.
Introduction
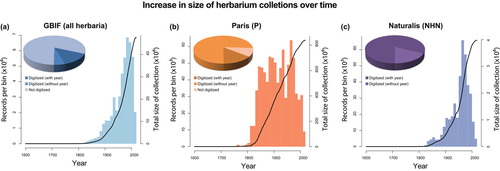
As invaluable botanical records of global plant diversity reaching into the sixteenth century (Sprague and Nelmes Citation1931), the combined collections of the world’s 3400 herbaria contain approximately 350 million samples spanning nearly 400 years (Soltis Citation2017) (Figure ). Within these collections, the specimens themselves can be thought of as direct records of species’ range and occurrences, their phenotypic variation and diseases, as well as time series of the phenological, demographic, and evolutionary changes and responses of populations to climatic variation, anthropogenic activity, and other biotic effects (Lister Citation2011). The digitization of herbarium specimen metadata has accelerated in recent years (Tulig et al. Citation2012), and the availability of this information through online repositories such as the Global Biodiversity Information Facility (GBIF) has made natural history collection more accessible to a diverse and general audience. This led to a remarkable expansion in the number and diversity of potential scientific applications for these records (Graham et al. Citation2004; Pyke and Ehrlich Citation2010; Soltis Citation2017).
Figure 1. Trends in the number of digitized plant specimens available in global herbaria. (a) Growth of specimen collections in global herbaria over time. All data on all “preserved specimens” in the kingdom Plantae were obtained from the Global Biodiversity Information Facility (GBIF), yielding 64.7 million records, of which 48.5 million (75%) contained metadata about the collection year. (b) Growth of the vascular plants collection (P) at the herbarium of the Muséum National d’Histoire Naturelle (MNHN, Paris) over time. Altogether, 5.4 million (90%) of the estimated 6 million specimens from P are databased in GBIF, of which 0.838 million (15%) contain information about the collection year. (c) Growth of the Naturalis Biodiversity Center (NHN, Netherlands) collections over time. All 4.8 million specimens from Naturalis are databased in GBIF, and 0.832 million (17%) of these contain no information about the collection year. In each panel, the bars show the number of specimens collected in each 10 year period, while the black line indicates the cumulative total number of collected specimens. The general trend shows an increasing rate of global specimen deposition starting from 1800, with very few specimens collected before that year. The growth rate of herbarium collections dropped markedly during the periods of World War I and World War II. During the last 30–40 years, the rate of specimen deposition has decreased, although it is not clear whether this reflects a real effect or a time lag or bias in digitization efforts. It is important to note that for many historical herbarium specimens, the precise collection date may be unknown, although often an approximate collection date can be gleaned from associated historical records (e.g. plants collected during a botanical expedition in 1804–1806). Therefore, although this information is missing in the GBIF database, those specimens could still be valuable in a study of temporal genetic variation.
Comparing modern genetic data with historical data obtained from herbarium specimens can offer unique insights into fundamental evolutionary questions. Although the utility of these collections for plant genetic studies was recognized early on (Rogers and Bendich Citation1985), researchers found that polymerase chain reaction (PCR) amplification yields were extremely variable between specimens (Savolainen et al. Citation1995). Fortunately, in recent decades there has been an increase in the accessibility of the genetic information stored in these specimens, mostly owing to advances in DNA extraction and sequencing approaches tailored for ancient DNA (aDNA), especially next-generation sequencing (NGS) (Pääbo et al. Citation2004; Yoshida et al. Citation2014). The evolution of genomes can now be directly observed without the need to infer the past from modern samples. This is especially relevant for endangered and extinct species, but also for invasive species (Staats et al. Citation2013), as the analysis of these species can lead to a better understanding of past biodiversity and adaptation processes.
In this article, the challenges of using old, degraded DNA from herbarium specimens are addressed along with examples illustrating how such analysis can be applied in evolutionary studies. We offer a brief review of novel usage cases of genetic and genomic sequencing in herbarium collections (Table ), and discuss the prospects for these approaches in the future. Throughout this article, we will refer to degraded DNA from historical herbarium specimens as aDNA, based on the presence of a detectable aDNA damage pattern, although we acknowledge that truly ancient plant specimens must be treated with additional stringency.
Table 1. Overview of genetic studies using herbarium samples.
Challenges and recent progress in using herbarium specimens in genetic studies
As is generally recognized for all natural history collections (Pyke and Ehrlich Citation2010), the use of herbarium specimens for ecological and evolutionary studies is limited by biases in the records themselves, including non-random and uneven collection over time and space (e.g. individual collectors contributing different taxa at different times); absences, errors, and inaccuracies in associated metadata (e.g. labels containing incorrect information on taxonomic identification or collection location); losses to flood and fire; or because particular specimens have been preferentially amassed or discarded by curators. Any study relying on genetic analysis of herbarium specimens should take care to ensure that the study questions can be answered despite these unavoidable biases.
The particular preservation methods used can influence the quality and accessibility of genetic information in herbarium specimens (Adams and Sharma Citation2010; Pyle and Adams Citation1989; Staats et al. Citation2011), although this general observation has not been subjected to systematic study, probably because the voucher preservation method is not always obvious. More recent collections are often deposited after a gentle drying process using warm air, paper, and a wooden press, historically, harsher methods (e.g. treatment of specimens with formalin, ethanol, and/or mercuric chloride) were more popular (Bakker Citation2015; Doyle and Dickson Citation1987; Nickrent Citation1994; Srinivasan, Sedmak, and Jewell Citation2002). In tropical fieldwork scenarios, high-temperature drying and treatment with ethanol and formaldehyde are often the only way to achieve dry, fungus-free plant specimens for mounting (Bakker Citation2015; Nickrent Citation1994).
It follows that the age of a specimen can strongly influence the accessibility of its genetic information. Weiß et al. (Citation2016) showed that this is particularly true for preserved plants via bioinformatic estimation that DNA in mounted herbarium specimens decays six times faster than in bone. Indeed, in comparison to more “ancient” samples, DNA extracted from historical herbarium specimens can be extremely fragmented in spite of their relatively young age (Adams and Sharma Citation2010; Staats et al. Citation2011). Therefore, precautions used for truly ancient samples have been called for even in the use of historical plant specimens (Shepherd and Perrie Citation2014). Since ancient specimens are often characterized by low DNA concentrations, attempts to amplify the endogenous DNA in PCR are prone to modern-day contamination. Thus, special care should be applied during sample handling, and pre-PCR steps should be performed in dedicated clean laboratories (Pääbo et al. Citation2004). Although some specimens are simply too degraded to be used for sequencing, new methods for enriching DNA extracts with extremely short (40–100 bp) DNA fragments (e.g. Gutaker et al. Citation2017) push the boundaries of what can be sequenced to very low-quality specimens containing only short fragments that are usually not recoverable with standard extraction protocols. For specimens too precious to be used for destructive sampling (e.g. holotypes), new non-destructive sampling approaches (e.g. Shepherd Citation2017) promise to make even these specimens available for genetic analysis.
Shortly after an organism’s death, the endogenous DNA begins to decay due to exposure to intracellular nucleases and microorganisms as well as hydrolytic and oxidative processes. These processes lead to a substantial reduction in mean DNA fragment length as well as characteristic aDNA damage patterns. Most prominently, at the single-stranded overhanging ends of double-stranded DNA fragments, the deamination of cytosine to uracil plays a major role, since uracil pairs with adenine while cytosine pairs with guanine during DNA amplification. During PCR, this leads to C to T transitions (or G to A, depending on the strand sequenced) due to nucleotide misincorporation by DNA polymerase (Dabney, Meyer, and Pääbo Citation2013; Staats et al. Citation2011). This characteristic damage pattern can be used to authenticate aDNA and has successfully been used to separate aDNA from modern contamination in studies of ancient hominins (Skoglund et al. Citation2014). However, damage patterns can also lead to problems, especially in direct PCR approaches, as primers may not bind to fragments with a high rate of C to T misincorporations. In addition, many of the short fragments found in the DNA extracts may not even cover the whole region of interest. Therefore, recovering the region of interest from aDNA is not always possible with direct PCR approaches. Moreover, the characteristic damage pattern is often masked by the primers during PCR as it occurs only at fragment ends. So, the damage pattern cannot be used to authenticate the recovered sequences (Gutaker and Burbano Citation2017).
In contrast, NGS approaches are better suited for aDNA since they are based on short fragments (modern samples are usually physically or enzymatically fragmented before NGS library preparation) (Gutaker and Burbano Citation2017; Yoshida, Sasaki, and Kamoun Citation2015). Adapters are ligated to the ends of the fragments, increasing the number of fragments used for sequencing in comparison with PCR approaches. Moreover, the fragment ends are sequenced, and the characteristic damage pattern can be used for authentication (Gutaker and Burbano Citation2017). However, DNA misincorporation can lead to problems in the downstream analysis since some identified single-nucleotide polymorphisms (SNPs) may only be present as a result of post-mortem DNA damage. To account for this problem, software tools such as mapDamage (Jónsson et al. Citation2013) can be used in the bioinformatic analysis of the sequencing reads. Base qualities are then recalibrated based on the damage pattern, reducing the probability of falsely called SNPs.
Mining the diversity of species genomes stored in herbaria
Herbarium collections make available a wealth of specimens that otherwise could not be obtained, or only via costly or difficult fieldwork, including rare specimens (e.g. types), endangered or extinct species, and geographically restricted populations (Shepherd and Perrie Citation2014). Destructive sampling of such specimens to provide DNA for use in molecular systematics studies is a well-established and now frequently used method (e.g. Koch et al. Citation2017; Lehtonen et al. Citation2010; Martin et al. Citation2018; Savolainen et al. Citation1995; Wurdack and Davis Citation2009). It is therefore possible to include herbarium specimens with rare morphotypes or from a wide range of sampling locations that would not easily be obtained through fieldwork (Olofsson et al. Citation2016).
Historical collections can also be used to study now-extinct species in a phylogenetic context. For example, Zedane et al. (Citation2016) used genome “skimming” data (low-depth shotgun sequencing) from an extinct, monotypic genus (Hesperelaea A.Gray, Oleaceae) that is only known from a single, 140-year-old collection, to estimate the phylogenetic position of the genus.
Newly developed spatial phylogenetic analysis methods (Mishler et al. Citation2014) can reveal surprising insights into the spatial distribution of plant genetic diversity, which can be used to objectively determine where to focus biodiversity conservation resources (e.g. Baldwin et al. Citation2017; Thornhill et al. Citation2016). These methods require a reference library of conserved DNA barcode sequences for every taxon above a particular taxonomic level occurring within a continent-scale region. To complete the initial sequence alignment, authors of these studies utilize traditional PCR and Sanger sequencing of herbarium specimens to generate the missing DNA barcode data required to complement massive data collections already available in public repositories such as GenBank (Benson et al. Citation2005).
High-throughput sequencing (NGS) approaches have already been shown to improve the accessibility of conserved genetic markers in degraded samples. For example, Prosser et al. (Citation2016) used a nested PCR approach and Ion Torrent™ Personal Genome Machine™ (Life Technologies) NGS to recover some full-length DNA sequences of the cytochrome oxidase I (COI) barcode from degraded type specimens of Geometridae (Lepidoptera) collected up to 120 years ago, with per-sample costs close to those of traditional Sanger sequencing. Although the authors utilized insect specimens, this approach can certainly be implemented with herbarium specimens as well. Hart et al. (Citation2016) used a target enrichment approach to retrieve hundreds of transcriptome-associated nuclear loci from herbarium specimens of the Neotropical genus Inga, reporting success even with extractions yielding extremely low masses (16 ng) of DNA. Such approaches could become particularly valuable for connecting type specimens to genetic sequences, and therefore help to resolve long-standing taxonomic issues (Liimatainen et al. Citation2014). Sánchez Barreiro et al. (Citation2017) used a modern genotyping-by-sequencing (GBS) data set to design RNA baits that were used to enrich for restriction enzyme-associated loci in historical herbarium specimens collected up to 180 years ago. The targeted loci were enriched by 19–151-fold in comparison with shotgun sequencing. This approach is useful for integrating historical, degraded samples into existing reduced representation library data sets for population genetic studies (Sánchez Barreiro et al. Citation2017).
It has also been shown that de novo assembly of genome-skimming data is a relatively straightforward approach by which to generate sequences of high-copy regions such as whole plastome sequences and nuclear ribosomal units (Bakker Citation2017; Besnard et al. Citation2014; Staats et al. Citation2013; Zedane et al. Citation2016). The ongoing PhyloNorway project uses a genome-skimming approach to assemble plastome sequences and aims to complete a reference database for all Norwegian vascular plants, with the majority of the material coming from herbarium collections (Taberlet et al. Citation2018). If a reference sequence is already available, even low-copy genes can be investigated using NGS data, enabling comparative genomics (Besnard et al. Citation2014). Moreover, “skimming” data from herbarium specimens can also be used to extract a large number of nuclear SNPs, which can be used to infer genetic structure (Olofsson et al. Citation2016).
In addition to improvements in recovering genetic data from historical herbarium specimens, new computational pipelines such as PALEOMIX (Schubert et al. Citation2014), EAGER (Peltzer et al. Citation2016), and ATLAS (Link et al. Citation2017) have been developed specifically for genomic data from degraded specimens. Those software tools automate the bioinformatic analyses (e.g. DNA damage base quality recalibration, target capture enrichment efficiency calculation) that are required before evolutionary inference can begin.
Using herbarium specimens to study evolution, population history, and invasion
When drawing conclusions about the evolutionary history of species, inferences are generally based on present-day patterns of variation. However, this approach may give an incomplete picture of population history. Major demographic events such as population turnovers or bottlenecks can erase most of the genetic variation that existed in the past (Leonardi et al. Citation2017; Palkopoulou et al. Citation2016). Modern distributions of alleles and genotypes can therefore differ significantly from past distributions. Simulation studies showed that this is especially the case when alleles are surfing on a migration wave (Edmonds, Lillie, and Cavalli-Sforza Citation2004; Klopfstein, Currat, and Excoffier Citation2006), as may be the case, for example, in expanding populations of an invasive plant. Sequence data from historical specimens provide direct insights into a genome’s past states and offer the power to observe recent evolution in action. This direct genetic evidence can help to reveal the true history of potentially beneficial alleles and therefore increase our understanding about why they experienced selection, especially when this is combined with other available data such as climate records (Lister Citation2011). For example, historical genetic data from Ipomoea batatas (L.) Lam. (Roullier et al. 2013) and Anacamptis palustris (Jacq.) R.M.Bateman, Pridgeon & M.W.Chase (Cozzolino et al. Citation2007) and metagenomic data from the plant pathogen Phytophthora infestans (Mont.) de Bary, derived from nineteenth-century Solanum tuberosum L. collections (Martin et al. Citation2013; Yoshida et al. Citation2013), showed changes in the distribution of haplotypes that were linked to anthropogenic activity.
For plant population geneticists, herbarium specimens offer a unique opportunity to directly observe allele frequencies and population structure in historical times, as well as changes in these metrics in relation to the present day. Since the specimens are usually well preserved and contain metadata such as the exact sampling location, the collection date, and information about the habitat, herbaria also provide information about historical geographic distributions (Chauvel et al. Citation2006). Including historical samples in studies of genetic diversity often leads to surprising results. For example, Saltonstall (Citation2002) sequenced chloroplast markers from historical herbarium specimens of the reed Phragmites australis (Cav.) Trin. Ex Steud., finding that a non-native genotype had completed a “cryptic invasion” of the north-eastern USA, with pre-1900 native genotypes being completely replaced by an invasive haplotype over a period of just 20 years. Roullier et al. (2013) used both modern and historical herbarium samples (collected between the seventeenth century and the early twentieth century) of sweet potato (Ipomoea batatas) and found that most modern samples from eastern Polynesia carry a chloroplast haplogroup not found in the historical dataset from that region. Their results support the hypothesis that later introductions of sweet potato have replaced the initial pre-Columbian introduced population. Martin et al. (Citation2014b) performed an analysis of common ragweed (Ambrosia artemisiifolia L.) from North American herbarium specimens and showed that the geographic boundaries between two genetic clusters had changed substantially over the last 140 years, in association with the expansion of European agriculture. Cozzolino et al. (Citation2007) compared plastid DNA intron haplotype frequencies in nearly 100 historical herbarium specimens of a rare orchid (Anacamptis palustris) to present-day populations from Italy, finding that human-mediated habitat loss precipitated the extinction of particular haplotypes of this endangered plant over the last century. Combining historical genetic data with records of human activity can therefore increase our understanding of how and why plant species spread or become extinct.
Inferring the history of alleles known to have been influenced by natural selection can help to elucidate why they are advantageous. A scenario in which this could be especially useful is in the study of exotic plants that underwent rapid adaptation either before (Vandepitte et al. Citation2014) or after (Hodgins and Rieseberg Citation2011) invasion, specifically with regard to the genes underlying traits such as improved growth and reproduction. As each introduction event can be seen as an independent natural local adaptation experiment, historical genetic information can provide insight into fundamental questions regarding the evolution of introduced plants. Exposito-Alonso et al. (Citation2018) report sequences of 27 historical genomes from herbarium specimens of Arabidopsis thaliana (L.) Heynh. belonging to a specific haplogroup (HPG1) that is the most abundant lineage since the 1860s and possibly one of the first lineages of A. thaliana that colonized North America. They discovered alleles that had risen to high frequency in modern populations and are connected to growth and life history traits. This may provide a path for future studies that directly investigate the rate and processes of adaptation in colonizing plants.
Just as the fossil record can be used to calibrate branch lengths and estimate divergence times in a Bayesian phylogenetic framework (Forest Citation2009), historical sequences from herbarium specimens can be advantageous in phylogenetic studies. This approach was validated using historical herbarium samples with known collection dates as tip calibration points to probe the evolutionary timescale of the potato pathogen Phytophthora infestans, relating the species’ diversification to the Spanish Conquest of Central and South America (Martin et al. Citation2014a; Yoshida et al. Citation2013). The same approach was used in a study of Arabidopsis thaliana to estimate the substitution rate and the time of the most recent common ancestor, which relates to the colonization time of the plant in North America (Exposito-Alonso et al. Citation2018).
Studying plant pathogens and plant–pathogen interactions
Aside from the specimen’s endogenous DNA, extractions from herbarium samples also contain metagenomic DNA from microorganisms that populated the sample (Yoshida, Sasaki, and Kamoun Citation2015). This is especially useful in plant pathogen research as it enables researchers to directly probe the genetics of past disease epidemics and to reconstruct population dynamics of the microorganisms (Yoshida et al. Citation2014). Raw reads can be mapped against a reference for the pathogen as well as the host plant. Therefore, plant–pathogen interactions can be studied through time (Yoshida, Sasaki, and Kamoun Citation2015).
Characterizing microbial communities of conspecific samples collected from various locations and times enables the origins and introduction pathways of plant pathogens to be studied. Moreover, the evolution and population dynamics of the pathogens can be inferred when they are found in different periods. Herbarium studies of the oomycete Phytophthora infestans, which causes late potato blight, showed that the genetic structure of the pathogen changed in Europe since its introduction during the nineteenth century, which triggered the Irish potato famine (Yoshida et al. Citation2014). These collection-based studies also demonstrate that although the nineteenth-century outbreak was caused by the HERB-1 lineage, the distinct but closely related US-1 lineage was responsible for the early twentieth-century global outbreak of late potato blight disease, completely replacing HERB-1 in Europe (Saville, Martin, and Ristaino Citation2016; Yoshida et al. Citation2013).
In an exploratory study that remains the only work of its kind, Schubert et al. (Citation2014) showed that the metagenomic communities can differ substantially among herbarium vouchers. In their comparison of three historical infected domestic potato (Solanum tuberosum) specimens, one from Belgium collected in 1845 contained mostly Gram-positive Actinobacteria, while two collected from Germany in the 1880s consisted almost entirely of Gram-negative Gammaproteobacteria. The microbial communities preserved in historical plant samples remain an underexplored area, especially as they can elucidate past disease epidemics.
Malmstrom et al. (Citation2007) used a reverse-transcriptase PCR approach to amplify and Sanger sequence barley yellow dwarf viruses from historical herbarium specimens of various California grasses (Poaceae) up to 100 years old. They detected phylogenetically diverse viruses on wild grasses as far back in time as 1917, with a strong association between California/Australia and France/Morocco. Their results suggested intercontinental virus dispersal mediated by humans in historical times and relate to the aforementioned studies of Phytophthora infestans, pointing to the importance of human trade and early colonial activities in facilitating the movement of plant pathogens between diverse localities.
Conclusion and future prospects
Methodological improvements in aDNA extraction and analysis now make it possible to access the vast number of plant genomes preserved in world herbaria. The genomic data can be used for diverse evolutionary analyses, including but not limited to estimating phylogenetic relationships and reconstructing population history. Comparisons of historical and modern genomic data can be used to identify genomic regions under selection, and further functional analysis of those regions makes it possible to identify the pathways that contribute most to adaptation, especially in introduced and invasive plants.
Elucidating the mechanisms underlying adaptation of recently introduced plant species will be a fundamental step in understanding how species respond to environmental change and anthropogenic disturbance. This is of particular interest considering the pressing threat of global climate change, and as large numbers of species are introduced to new habitats. To understand how species have responded to these processes in the past, future studies should combine genetic and climatic data with metadata from voucher labels (e.g. information about the habitat) and morphological metrics taken directly from the pressed plants. For species represented by sufficiently abundant collections, it will be possible to infer temporal changes in genetic diversity by grouping conspecific specimens (e.g. by year of collection and/or sampling location) and then applying population genetic approaches. Thus, predictions about future responses to environmental change can be based on direct measurements from the historical collections.
Finally, studying the history of interactions of plant pathogens with their hosts will increase the understanding of epidemics and inform the management of future outbreaks. Novel insights could be gained from metagenomic analysis of herbarium specimen DNA, which can reveal the presence and genotypes of pathogens associated with specimens collected in the past, some of which may even be unrelated to known disease epidemics.
Disclosure statement
No potential conflict of interest was reported by the authors.
Notes on contributors
Vanessa C. Bieker is a PhD student in the Department of Natural History at the NTNU University Museum, Trondheim. Her research projects focus on the use of (historical) herbarium specimens to elucidate the evolutionary genomics of plant invasion. Contribution: Co-wrote the paper.
Michael D. Martin is an associate professor in the Department of Natural History at the NTNU University Museum, Trondheim. His research focuses on the use of museum collections to investigate the evolutionary history of natural populations. Contribution: Co-wrote the paper.
Related Research Data
References
- Adams, R. P. , and L. N. Sharma . 2010. “DNA from Herbarium Specimens: I. Correlation of DNA Size with Specimen Age.” Phytologia 92: 346–353.
- Ames, M. , and D. M. Spooner . 2008. “DNA from Herbarium Specimens Settles a Controversy about Origins of the European Potato.” American Journal of Botany 95 (2): 252–257.10.3732/ajb.95.2.252
- Bakker, F. T. 2015. “DNA Sequences from Plant Herbarium Tissue.” In Next-Generation Sequencing in Plant Systematics , edited by E. Hörandl and M. S. Appelhans , 1–18. Königstein, Germany: Koeltz Scientific Books.
- Bakker, F. T. 2017. “Herbarium Genomics: Skimming and Plastomics from Archival Specimens.” Webbia 72: 35–45.10.1080/00837792.2017.1313383
- Bakker, F. T. , D. Lei , J. Yu , S. Mohammadin , Z. Wei , S. van de Kerke , B. Gravendeel , et al . 2016. “Herbarium Genomics: Plastome Sequence Assembly from a Range of Herbarium Specimens Using an Iterative Organelle Genome Assembly Pipeline.” Biological Journal of the Linnean Society 117: 33–43.10.1111/bij.12642
- Baldwin, B. G. , A. H. Thornhill , W. A. Freyman , D. D. Ackerly , M. M. Kling , N. Morueta-Holme , and B. D. Mishler . 2017. “Species Richness and Endemism in the Native Flora of California.” American Journal of Botany 104: 487–501.10.3732/ajb.1600326
- Benson, D. A. , I. Karsch-Mizrachi , D. J. Lipman , J. Ostell , and D. L. Wheeler . 2005. “GenBank.” Nucleic Acids Research 33: D34–D38.
- Besnard, G. , P.-A. Christin , P.-J. G. Malé , E. Lhuillier , C. Lauzeral , E. Coissac , and M. S. Vorontsova . 2014. “From Museums to Genomics: Old Herbarium Specimens Shed Light on a C3 to C4 Transition.” Journal of Experimental Botany 65: 6711–6721.10.1093/jxb/eru395
- Chauvel, B. , F. Dessaint , C. Cardinal-Legrand , and F. Bretagnolle . 2006. “The Historical Spread of Ambrosia artemisiifolia L. in France from Herbarium Records.” Journal of Biogeography 33: 665–673.10.1111/jbi.2006.33.issue-4
- Cozzolino, S. , D. Cafasso , G. Pellegrino , A. Musacchio , and A. Widmer . 2007. “Genetic Variation in Time and Space: The Use of Herbarium Specimens to Reconstruct Patterns of Genetic Variation in the Endangered Orchid Anacamptis palustris .” Conservation Genetics 8: 629–639.10.1007/s10592-006-9209-7
- Dabney, J. , M. Meyer , and S. Pääbo . 2013. “Ancient DNA Damage.” Cold Spring Harbor Perspectives in Biology 5: a012567.
- Doyle, J. J. , and E. E. Dickson . 1987. “Preservation of Plant Samples for DNA Restriction Endonuclease Analysis.” Taxon 36: 715–722.10.2307/1221122
- Edmonds, C. A. , A. S. Lillie , and L. L. Cavalli-Sforza . 2004. “Mutations Arising in the Wave Front of an Expanding Population.” Proceedings of the National Academy of Sciences 101: 975–979.10.1073/pnas.0308064100
- Exposito-Alonso, M. , C. Becker , V. J. Schuenemann , E. Reiter , C. Setzer , R. Slovak , B. Brachi , et al . 2018. “The Rate and Potential Relevance of New Mutations in a Colonizing Plant Lineage.” PLoS Genetics 14(2): e1007155. doi:10.1371/journal.pgen.1007155.
- Forest, F. 2009. “Calibrating the Tree of Life: Fossils, Molecules and Evolutionary Timescales.” Annals of Botany 104: 789–794.10.1093/aob/mcp192
- Graham, C. H. , S. Ferrier , F. Huettman , C. Moritz , and A. T. Peterson . 2004. “New Developments in Museum-Based Informatics and Applications in Biodiversity Analysis.” Trends in Ecology and Evolution 19: 497–503.10.1016/j.tree.2004.07.006
- Gutaker, R. M. , and H. A. Burbano . 2017. “Reinforcing Plant Evolutionary Genomics Using Ancient DNA.” Current Opinion in Plant Biology 36: 38–45.10.1016/j.pbi.2017.01.002
- Gutaker, R. M. , E. Reiter , A. Furtwängler , V. J. Schuenemann , and H. A. Burbano . 2017. “Extraction of Ultrashort DNA Molecules from Herbarium Specimens.” BioTechniques 62: 76–79.
- Hart, M. L. , L. L. Forrest , J. A. Nicholls , and C. A. Kidner . 2016. “Retrieval of Hundreds of Nuclear Loci from Herbarium Specimens.” Taxon 65: 1081–1092.10.12705/655.9
- Hodgins, K. A. , and L. Rieseberg . 2011. “Genetic Differentiation in Life-History Traits of Introduced and Native Common Ragweed (Ambrosia artemisiifolia) Populations.” Journal of Evolutionary Biology 24: 2731–2749.10.1111/jeb.2011.24.issue-12
- Hughey, J. R. , P. W. Gabrielson , L. Rohmer , J. Tortolani , M. Silva , K. A. Miller , J. D. Young , C. Martell , and E. Ruediger . 2014. “Minimally Destructive Sampling of Type Specimens of Pyropia (Bangiales, Rhodophyta) Recovers Complete Plastid and Mitochondrial Genomes.” Scientific Reports 4: 1–10.
- Jónsson, H. , A. Ginolhac , M. Schubert , P. L. F. Johnson , and L. Orlando . 2013. “mapDamage2.0: Fast Approximate Bayesian Estimates of Ancient DNA Damage Parameters.” Bioinformatics 29: 1682–1684.10.1093/bioinformatics/btt193
- Klopfstein, S. , M. Currat , and L. Excoffier . 2006. “The Fate of Mutations Surfing on the Wave of a Range Expansion.” Molecular Biology and Evolution 23: 482–490.10.1093/molbev/msj057
- Koch, M. A. , F. Michling , A. Walther , X. C. Huang , L. Tewes , and C. Müller . 2017. “Early-Mid Pleistocene Genetic Differentiation and Range Expansions as Exemplified by Invasive Eurasian Bunias orientalis (Brassicaceae) Indicates the Caucasus as Key Region.” Scientific Reports 7: 8018.10.1038/s41598-017-17085-8
- Lehtonen, S. , H. Tuomisto , G. Rouhan , and M. J. M. Christenhusz . 2010. “Phylogenetics and Classification of the Pantropical Fern Family Lindsaeaceae.” Botanical Journal of the Linnean Society 163: 305–359.10.1111/boj.2010.163.issue-3
- Leonardi, M. , P. Librado , C. Der Sarkissian , M. Schubert , A. H. Alfarhan , S. A. Alquraishi , K. A. S. Al-Rasheid , C. Gamba , E. Willerslev , and L. Orlando . 2017. “Evolutionary Patterns and Processes: Lessons from Ancient DNA.” Systematic Biology 66: e1–e29.
- Liimatainen, K. , T. Niskanen , B. Dima , I. Kytövuori , J. F. Ammirati , and T. G. Frøslev . 2014. “The Largest Type Study of Agaricales Species to Date: Bringing Identification and Nomenclature of Phlegmacium (Cortinarius) into the DNA Era.” Persoonia 33: 98–140.10.3767/003158514X684681
- Link, V. , A. Kousathanas , K. Veeramah , C. Sell , A. Scheu , and D. Wegmann . 2017. “ATLAS: Analysis Tools for Low-Depth and Ancient Samples.” bioRxiv 105346. doi:10.1101/105346.
- Lister, A. M. 2011. “Natural History Collections as Sources of Long-Term Datasets.” Trends in Ecology and Evolution 26: 153–154.10.1016/j.tree.2010.12.009
- Malmstrom, C. M. , R. Shu , E. W. Linton , L. A. Newton , and M. A. Cook . 2007. “Barley Yellow Dwarf Viruses (BYDVs) Preserved in Herbarium Specimens Illuminate Historical Disease Ecology of Invasive and Native Grasses.” Journal of Ecology 95: 1153–1166.10.1111/jec.2007.95.issue-6
- Martin, M. D. , E. Capellini , J. A. Samaniego , M. L. Zepeda , P. F. Campos , A. Seguin-Orlando , N. Wales , et al . 2013. “Reconstructing Genome Evolution in Historic Samples of the Irish Potato Famine Pathogen.” Nature Communications 4: 2172.
- Martin, M. D. , S. Y. W. Ho , N. Wales , J. B. Ristaino , and M. T. P. Gilbert . 2014a. “Persistence of the Mitochondrial Lineage Responsible for the Irish Potato Famine in Extant New World Phytophthora infestans .” Molecular Biology and Evolution 31: 1414–1420.10.1093/molbev/msu086
- Martin, M. D. , E. A. Zimmer , M. T. Olsen , A. D. Foote , M. T. P. Gilbert , and G. S. Brush . 2014b. “Herbarium Specimens Reveal a Historical Shift in Phylogeographic Structure of Common Ragweed during Native Range Disturbance.” Molecular Ecology 23: 1701–1716.10.1111/mec.12675
- Martin, M. D. , E. Quiroz-Claros , G. S. Brush , and E. A. Zimmer . 2018. “Herbarium Collection-Based Phylogenetics of the Ragweeds (Ambrosia artemisiifolia L.)” Molecular Phylogenetics and Evolution 120: 335–341.10.1016/j.ympev.2017.12.023
- Mishler, B. D. , N. Knerr , C. E. González-Orozco , A. H. Thornhill , S. W. Laffan , and J. T. Miller . 2014. “Phylogenetic Measures of Biodiversity and Neo- and Paleo-endemism in Australian Acacia .” Nature Communications 5: 4473.
- Nickrent, D. L. 1994. “From Field to Film: Rapid Sequencing Methods for Field-Collected Plant Species.” BioTechniques 16: 470–475.
- Olofsson, J. K. , M. Bianconi , G. Besnard , L. T. Dunning , M. R. Lundgren , H. Holota , M. S. Vorontsova , et al . 2016. “Genome Biogeography Reveals the Intraspecific Spread of Adaptive Mutations for a Complex Trait.” Molecular Ecology 25 (24): 6107–6123.10.1111/mec.2016.25.issue-24
- Pääbo, S. , H. Poinar , D. Serre , V. Jaenicke-Després , J. Hebler , N. Rohland , M. Kuch , J. Krause , L. Vigilant , and M. Hofreiter . 2004. “Genetic Analyses from Ancient DNA.” Annual Review of Genetics 38: 645–679.10.1146/annurev.genet.37.110801.143214
- Palkopoulou, E. , M. Baca , N. I. Abramson , M. Sablin , P. Socha , A. Nadachowski , S. Prost , et al . 2016. “Synchronous Genetic Turnovers across Western Eurasia in Late Pleistocene Collared Lemmings.” Global Change Biology 22: 1710–1721.10.1111/gcb.13214
- Peltzer, A. , G. Jäger , A. Herbig , A. Seitz , C. Kniep , J. Krause , and K. Nieselt . 2016. “EAGER: Efficient Ancient Genome Reconstruction.” Genome Biology 17: 60.
- Prosser, S. W. J. , J. R. deWaard , S. E. Miller , and P. D. N. Hebert . 2016. “DNA Barcodes from Century-Old Type Specimens Using Next-Generation Sequencing.” Molecular Ecology Resources 16: 487–497.10.1111/men.2016.16.issue-2
- Pyke, G. H. , and P. R. Ehrlich . 2010. “Biological Collections and Ecological/Environmental Research: A Review, Some Observations and a Look to the Future.” Biological Reviews 85: 247–266.10.1111/brv.2010.85.issue-2
- Pyle, M. M. , and R. P. Adams . 1989. “ In Situ Preservation of DNA in Plant Specimens.” Taxon 38: 576–581.10.2307/1222632
- Rogers, S. O. , and A. J. Bendich . 1985. “Extraction of DNA from Milligram Amounts of Fresh, Herbarium and Mummified Plant Tissues.” Plant Molecular Biology 5: 69–76.10.1007/BF00020088
- Roullier, C. , L. Benoit , D. B. McKey , and V. Lebot . 2013. “Historical Collections Reveal Patterns of Diffusion of Sweet Potato in Oceania Obscured by Modern Plant Movements and Recombination.” Proceedings of the National Academy of Sciences 110: 2205–2210.10.1073/pnas.1211049110
- Saltonstall, K. 2002. “Cryptic Invasion by a Non-Native Genotype of the Common Reed, Phragmites australis, into North America.” Proceedings of the National Academy of Sciences of the United States of America 99: 2445–2449.10.1073/pnas.032477999
- Sánchez Barreiro, F. , F. G. Vieira , M. D. Martin , J. Haile , M. T. P. Gilbert , and N. Wales . 2017. “Characterizing Restriction Enzyme-Associated Loci in Historic Ragweed (Ambrosia artemisiifolia) Voucher Specimens Using Custom-Designed RNA Probes.” Molecular Ecology Resources 17: 209–220.10.1111/men.2017.17.issue-2
- Saville, A. C. , M. D. Martin , and J. B. Ristaino . 2016. “Historic Late Blight Outbreaks Caused by a Widespread Dominant Lineage of Phytophthora infestans (Mont.) de Bary.” PLoS One 11: e0168381.10.1371/journal.pone.0168381
- Savolainen, V. , P. Cuénoud , R. Spichiger , M. D. P. Martinez , M. Crèvecoeur , and J. Manen . 1995. “The Use of Herbarium Specimens in DNA Phylogenetics: Evaluation and Improvement.” Plant Systematics and Evolution 197: 87–98.10.1007/BF00984634
- Schaefer, H. , and S. S. Renner . 2010. “A Gift from the New World? The West African Crop Cucumeropsis mannii and the American Posadaea sphaerocarpa (Cucurbitaceae) Are the Same Species.” Systematic Botany 35: 534–540.10.1600/036364410792495818
- Schubert, M. , L. Ermini , C. Der Sarkissian , H. Jónsson , A. Ginolhac , R. Schaefer , M. D. Martin , et al . 2014. “Characterization of Ancient and Modern Genomes by SNP Detection and Phylogenomic and Metagenomic Analysis Using PALEOMIX.” Nature Protocols 9: 1056–1082.10.1038/nprot.2014.063
- Sebastian, P. , H. Schaefer , and S. S. Renner . 2010. “Darwin’s Galapagos Gourd: Providing New Insights 175 Years after His Visit.” J. Biogeogr 37: 975–978.
- Shepherd, L. D. 2017. “A Non-Destructive DNA Sampling Technique for Herbarium Specimens.” PLoS One 12: e0183555.10.1371/journal.pone.0183555
- Shepherd, L. , and L. Perrie . 2014. “Genetic Analyses of Herbarium Material: Is More Care Required?” Taxon 63: 972–973.10.12705/635.2
- Skoglund, P. , B. H. Northoff , M. V. Shunkov , A. P. Derevianko , S. Pääbo , J. Krause , and M. Jakobsson . 2014. “Separating Endogenous Ancient DNA from Modern Day Contamination in a Siberian Neandertal.” Proceedings of the National Academy of Sciences of the United States of America 111: 2229–2234.10.1073/pnas.1318934111
- Soltis, P. S. 2017. “Digitization of Herbaria Enables Novel Research.” American Journal of Botany 104: 1–4.
- Sprague, T. A. , and E. Nelmes . 1931. “The Herbal of Leonhart Fuchs.” Botanical Journal of the Linnean Society 48: 545–642.10.1111/j.1095-8339.1931.tb00596.x
- Srinivasan, M. , D. Sedmak , and S. Jewell . 2002. “Effect of Fixatives and Tissue Processing on the Content and Integrity of Nucleic Acids.” The American Journal of Pathology 161: 1961–1971.10.1016/S0002-9440(10)64472-0
- Staats, M. , A. Cuenca , J. E. Richardson , R. Vrielink-van Ginkel , G. Petersen , O. Seberg , and F. T. Bakker . 2011. “DNA Damage in Plant Herbarium Tissue.” PLoS One 6: e28448.10.1371/journal.pone.0028448
- Staats, M. , R. H. J. Erkens , B. van de Vossenberg , J. J. Wieringa , K. Kraaijeveld , B. Stielow , J. Geml , J. E. Richardson , and F. T. Bakker . 2013. “Genomic Treasure Troves: Complete Genome Sequencing of Herbarium and Insect Museum Specimens.” PLoS One 8: e69189.10.1371/journal.pone.0069189
- Taberlet, P. , A. Bonin , L. Zinger , and E. Coissac . 2018. Environmental DNA for Biodiversity Research and Monitoring . Oxford: Oxford University Press.
- Thornhill, A. H. , B. D. Mishler , N. J. Knerr , C. E. González-Orozco , C. M. Costion , D. M. Crayn , S. W. Laffan , and J. T. Miller . 2016. “Continental-Scale Spatial Phylogenetics of Australian Angiosperms Provides Insights into Ecology, Evolution and Conservation.” Journal of Biogeography 43: 2085–2098.
- Tulig, M. , N. Tarnowsky , M. Bevans , A. Kirchgessner , and B. M. Thiers . 2012. “Increasing the Efficiency of Digitization Workflows for Herbarium Specimens.” ZooKeys 209: 103–113.10.3897/zookeys.209.3125
- Vandepitte, K. , T. de Meyer , K. Helsen , K. van Acker , I. Roldán-Ruiz , J. Mergeay , and O. Honnay . 2014. “Rapid Genetic Adaptation Precedes the Spread of an Exotic Plant Species.” Molecular Ecology 23: 2157–2164.10.1111/mec.12683
- Weiß, C. L. , V. J. Schuenemann , J. Devos , G. Shirsekar , E. Reiter , B. A. Gould , J. R. Stinchcombe , J. Krause , and H. A. Burbano . 2016. “Temporal Patterns of Damage and Decay Kinetics of DNA Retrieved from Plant Herbarium Specimens.” Royal Society Open Science 3: 160239.
- Wurdack, K. J. , and C. C. Davis . 2009. “Malpighiales Phylogenetics: Gaining Ground on One of the Most Recalcitrant Clades in the Angiosperm Tree of Life.” American Journal of Botany 96: 1551–1570.10.3732/ajb.0800207
- Yoshida, K. , V. J. Schuenemann , L. M. Cano , M. Pais , B. Mishra , R. Sharma , C. Lanz , et al. 2013. “The Rise and Fall of the Phytophthora infestans Lineage that Triggered the Irish Potato Famine.” eLife 2: e00731.
- Yoshida, K. , H. A. Burbano , J. Krause , M. Thines , D. Weigel , and S. Kamoun . 2014. “Mining Herbaria for Plant Pathogen Genomes: Back to the Future.” PLoS Pathogens 10: e1004028.10.1371/journal.ppat.1004028
- Yoshida, K. , E. Sasaki , and S. Kamoun . 2015. “Computational Analyses of Ancient Pathogen DNA from Herbarium Samples: Challenges and Prospects.” Frontiers in Plant Science 6: 771.
- Zedane, L. , C. Hong-Wa , J. Murienne , C. Jeziorski , B. G. Baldwin , and G. Besnard . 2016. “Museomics Illuminate the History of an Extinct, Paleoendemic Plant Lineage (Hesperelaea, Oleaceae) Known from an 1875 Collection from Guadalupe Island, Mexico.” Biological Journal of the Linnean Society 117: 44–57.10.1111/bij.12509