
In theory, scientific discovery follows the path of putting forward a hypothesis, testing it and either accepting it as a theory or discarding it based on the outcome of testing. As it often happens, real life is more complicated than this straightforward scheme; research undergoes unexpected twists and turns, and the total body of experimental evidence often both provides support for and refutes the hypothesis in question. After protracted arguments, the focus of scientific debate eventually shifts, often without solidifying the outcome in a clearly articulated conclusion allowing each side to cling to its own subset of gathered evidence. As a consequence, old misconceptions often persist in the literature, confusing newcomers and slowing the pace of scientific discovery. In this editorial, I identify some of these outdated concepts and confusions as they relate to mtDNA, outline their historical contexts and provide criticisms.
Attribution of mtDNA coding sequences and promoters to the H- or L-strand is inconsistent in the literature
Typically, the two strands of mtDNA differ in their nucleotide composition and therefore can be separated in denaturing buoyant density gradients of caesium chloride based on their G + T content (Vinograd et al. Citation1963). Indeed, guanine and thymine bases are ionized in alkaline solutions, and as a result, the DNA strand with the higher content of these bases becomes denser in alkaline solutions due to the subsequent acquisition of caesium ions (Vinograd et al. Citation1963; Wells and Larson Citation1972). In this way, a heavy (high G + T content) and light (low G + T content) strand can be identified in most mitochondrial genomes (the H-strand and L-strand, respectively). This distinction between the two mtDNA strands is associated with the first common confusion.
Today, the textbook definition of the coding DNA strand is the one that is identical in sequence to the RNA transcript (making accommodations for the exon/intron structure and for the fact that DNA contains thymine instead of uracil). When the first mtDNA genomes of humans and mice were sequenced in 1981 by Siv Anderson and colleagues and David Clayton’s group, respectively, the two groups used different definitions of the coding DNA strand (Anderson et al. Citation1981; Bibb et al. Citation1981). The Clayton’s group paper states: ‘The major coding strand of mouse mitochondrial DNA is the heavy (H) strand; most transcripts are therefore of light (L)- strand sequence and are complementary to the H strand. The sequence shown is L-strand sequence’ (Bibb et al. Citation1981). Therefore, this group used a definition of the coding strand which is incorrect by present-day standards. As a result, even though the human and mouse mitochondrial genomes have an identical organization, this difference in definitions led to opposite conclusions with respect to whether the H- or L-strand of mtDNA is the main coding strand. Guiseppe Attardi’s group also adopted Clayton’s definition. Collectively, Clayton’s and Attardi’s schools elucidated much of what we know today about mtDNA transcription and replication. Therefore, their terminology, while erroneous by the present-day standards, became widely accepted, and today, statements about how the H-strand encodes most genes are fairly common and can even be found in textbooks (Goodman Citation2007; Strachan and Read Citation2010). The latter fact ensures that the confusion will be perpetuated. Importantly, however, Barroso Lima and Prosdocimi examined 4205 vertebrate mitogenomes and established that in 4200 cases, the L-strand was the main coding strand (Barroso Lima and Prosdocimi Citation2018).
While the issue of whether the H- or L-strand of mtDNA is the main coding strand may appear somewhat esoteric, the underlying problem (i.e., inconsistent terminology) has tangible consequences in terms of mitochondrial promoter nomenclature. Indeed, promoter names typically align with the names of transcripts encoded in the sense strand and therefore associate with the sense/coding strand, whereas mitochondrial promoter names align with transcribed/antisense/noncoding strand. Perhaps even more confusing, transcripts from the light-strand promoter (LSP) which, for conventionally trained molecular biologists, suggest them being of the L-strand sequence, in the current paradigm prime replication of the H-strand (since the primer and mtDNA strand primed by it are contiguous, L-strand sequences cannot be prime extension of the H-strand).
mtDNA maps are inconsistently annotated in the literature
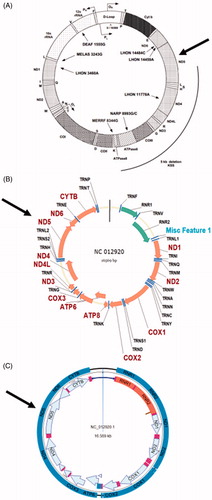
Another example of a frequent departure of the mitochondrial field from the existing molecular biology conventions can be seen in the presentation of circular maps of mtDNA, especially in biomedical studies. Whereas conventionally, circular genomes are annotated clockwise, many labs still follow Clayton’s counterclockwise annotation (Bibb et al. Citation1981; Falkenberg et al. Citation2007). The confusion is further confounded by the fact that the defiance of existing conventions is never acknowledged. Oftentimes, this results in an apparent disconnect between a map and sequence from the same source. For example, the MITOMAP database provides a map, an annotation and the reference sequence for human mtDNA. The map provided there (https://mitomap.org/foswiki/pub/MITOMAP/MitomapFigures/mitomapgenome.pdf) is a classical Clayton counterclockwise map (), whereas maps automatically generated from the reference sequence provided by the same source using software packages such as VectorNTI, DNASTAR or UGENE all have conventional clockwise annotations ().
Figure 1. mtDNA maps as A, found on MITOMAP website (https://mitomap.org/foswiki/pub/MITOMAP/MitomapFigures/mitomapgenome.pdf), or automatically generated from GenBank entry NC_012920 (revised Cambridge Reference Sequence) by Vector NTI (B) or DNAStar (C) software packages from GenBank entry NC_012920, referenced on the MITOMAP website. For orientation, arrows indicate location of the MT-ND5 gene.
Misconceptions regarding mtDNA mutagenesis and the contribution of reactive oxygen species to this process continue to persist
It is generally accepted that mtDNA accumulates mutations at a higher rate than nuclear DNA, and early attempts to rationalize this disparity are associated with a set of speculations, which are often presented as the established facts in introductory sections of manuscripts. This subject has been reviewed previously (Alexeyev Citation2009; Alexeyev et al. Citation2013; Kauppila and Stewart Citation2015), so here I will only reiterate the salient points and provide rebuttals.
Speculation#1. mtDNA mutagenesis results from reactive oxygen species (ROS) damage because mtDNA is located in close proximity to a major cellular source of ROS.
–While mitochondria are indeed a major cellular source of ROS, it is unclear whether mtDNA is freely accessible to mitochondrial ROS. Studies show that mtDNA mutations lack the canonical ROS signature (G > T transversions) (Kennedy et al. Citation2013; Ju et al. Citation2014; Itsara et al. Citation2014). The lack of such a signature is inconsistent with the notion of the leading role of ROS in mtDNA mutagenesis. Therefore, the contribution of mtDNA's subcellular location to mutagenesis is speculative.
Speculation #2. mtDNA is more susceptible to mutagenesis because it lacks ‘protective’ histones.
–Since it is impossible to generate cells that do not express histones, the protective role of histones has been impossible to elucidate directly in vivo. In vitro, histones can act as either sensitizers or be protective, depending on the experimental system used. It has also been shown that mitochondrial nucleoid proteins are as protective as histones (reviewed in Alexeyev et al. (Citation2013)). Therefore, the notion of the ‘protective role’ of histones is speculation rather than fact.
Speculation #3. mtDNA mutates at higher rates because mitochondria possess a reduced complement of DNA repair pathways.
–Mitochondria are proficient in the base excision repair (BER) pathway, the main pathway to repair oxidative and some forms of alkylating DNA damage in both the nucleus and mitochondria. Evidence suggests that oxidative damage in mtDNA may be repaired even more efficiently than in the nucleus (Thorslund et al. Citation2002). mtDNA damage that cannot be repaired can be addressed by degradation and resynthesis of the damaged molecule (Shokolenko et al. Citation2009; Alexeyev et al. Citation2013). The author is not aware of credible experimental evidence to support the notion that mtDNA mutation rates can be reduced by expanding the repertoire of DNA repair pathways available in mitochondria. While it is plausible that the lack of e.g. nucleotide excision repair (NER) or mismatch repair (MMR) pathways in mitochondria may contribute to elevated rates of mtDNA mutagenesis, this notion is not supported by experimental evidence. Importantly, most substrates for BER and MMR do not induce replicative DNA base mispairing. Rather, they induce mutations due to the low fidelity of DNA polymerases involved in the repair or bypass of these lesions in the nucleus. As a result, the lack of NER and MMR pathways in mitochondria may play a protective role, as counterintuitive as it may sound. Indeed, in the absence of NER, damaged mtDNA molecules are presumed to be channelled for degradation (Shokolenko et al. Citation2014), thus avoiding the error-prone repair afforded by NER. Therefore, the notion that mtDNA mutagenesis is driven primarily by the inability to repair certain types of DNA damage is speculative.
Speculation #4. mtDNA mutagenesis is driven by a ‘vicious cycle’ of DNA damage and ROS production.
–The ‘vicious cycle’ is a hypothetical construct that rose to notoriety with the mitochondrial theory of aging. It is based on the assumption that the majority of mtDNA mutations lead to mitochondrial dysfunction, which is necessarily accompanied by increased ROS production, which in turn leads to new mtDNA mutations, etc., in a feed-forward ‘vicious’ cycle. This concept was refuted by observations that a dramatic (several fold higher than can be observed under physiological conditions) accumulation of mtDNA mutations in mice due to a proofreading defect in PolG is not accompanied by increased oxidative stress (Trifunovic et al. Citation2005). Also, see the rebuttal for Speculation #1.
Conclusions
The current status quo with respect to the acceptance of inconsistent terminologies, variable annotation of mtDNA maps and the use of outdated concepts is untenable. The realignment of the mtDNA field with mainstream molecular biology would require a concerted and sustained effort on the part of the research community. The first step on this road is clearly identifying the existing confusions and misconceptions, which I hope to accomplish with this editorial.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Alexeyev MF. 2009. Is there more to aging than mitochondrial DNA and reactive oxygen species? FEBS J. 276(20):5768–5787.
- Alexeyev M, Shokolenko I, Wilson G, LeDoux S. 2013. The maintenance of mitochondrial DNA integrity–critical analysis and update. Cold Spring Harbor Perspect Biol. 5(5):a012641–a012641.
- Anderson S, Bankier AT, Barrell BG, de Bruijn MHL, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, et al. 1981. Sequence and organization of the human mitochondrial genome. Nature. 290(5806):457–465.
- Barroso Lima NC, Prosdocimi F. 2018. The heavy strand dilemma of vertebrate mitochondria on genome sequencing age: number of encoded genes or G + T content? Mitochondrial DNA Part A DNA Mapp Sequencing Anal. 29(2):300–302.
- Bibb MJ, Van Etten RA, Wright CT, Walberg MW, Clayton DA. 1981. Sequence and gene organization of mouse mitochondrial DNA. Cell. 26(2):167–180.
- Falkenberg M, Larsson NG, Gustafsson CM. 2007. DNA replication and transcription in mammalian mitochondria. Annu Rev Biochem. 76(1):679–699.
- Goodman SR. 2007. Medical Cell Biology. 3rd ed. Amsterdam: Academic Press.
- Itsara LS, Kennedy SR, Fox EJ, Yu S, Hewitt JJ, Sanchez-Contreras M, Cardozo-Pelaez F, Pallanck LJ. 2014. Oxidative stress is not a major contributor to somatic mitochondrial DNA mutations. PLoS Genet. 10(2):e1003974.
- Ju YS, Alexandrov LB, Gerstung M, Martincorena I, Nik-Zainal S, Ramakrishna M, Davies HR, Papaemmanuil E, Gundem G, Shlien A, et al. 2014. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife. 3:e02935.
- Kauppila JH, Stewart JB. 2015. Mitochondrial DNA: radically free of free-radical driven mutations. Biochim Biophys Acta. 1847(11):1354–1361.
- Kennedy SR, Salk JJ, Schmitt MW, Loeb LA. 2013. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 9(9):e1003794.
- Shokolenko I, Venediktova N, Bochkareva A, Wilson GL, Alexeyev MF. 2009. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 37(8):2539–2548.
- Shokolenko IN, Wilson GL, Alexeyev MF. 2014. Aging: a mitochondrial DNA perspective, critical analysis and an update. WJEM. 4(4):46–57.
- Strachan T, Read A. 2010. Human molecular genetics. 4th ed. New York (NY): Garland Science.
- Thorslund T, Sunesen M, Bohr VA, Stevnsner T. 2002. Repair of 8-oxoG is slower in endogenous nuclear genes than in mitochondrial DNA and is without strand bias. DNA Repair. 1(4):261–273.
- Trifunovic A, Hansson A, Wredenberg A, Rovio AT, Dufour E, Khvorostov I, Spelbrink JN, Wibom R, Jacobs HT, Larsson NG. 2005. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc Natl Acad Sci U S A. 102(50):17993–17998.
- Vinograd J, Morris J, Davidson N, Dove WF. Jr. 1963. The bouyant behavior of viral and bacterial DNA in alkaline CsCl. Proc Natl Acad Sci U S A. 49(1):12–17.
- Wells RD, Larson JE. 1972. Buoyant density studies on natural and synthetic deoxyribonucleic acids in neutral and alkaline solutions. J Biol Chem. 247(11):3405–3409.