
Abstract
We reevaluate the treatment of mercury poisoning, incorporating recent advances in understanding of mercury toxicity and the mercury:selenium interaction. This review focuses on: 1) the role, limitations and benefits of chelation (Unithiol, succimer and N-Acetylcysteine); 2) the role of selenium supplementation; and 3) how the different forms of mercury are impacted by use of chelation and selenium. Unithiol and succimer produce increases in urinary excretion of mercury and to a lesser degree blood and total body mercury. The primary role of N-acetylcysteine is increasing renal mercury excretion, similar to the thiol-chelators. Additional unique features of acetylcysteine include increased efflux of methylmercury from the brain, and reduced oxidative stress via increased glutathione production. The role of selenium includes: 1) restoration of selenoprotein activity, 2) protection against mitochondrial injury and DNA damage, 3) demethylation of methylmercury, 4) sequestering of mercury via Hg:Se complexes, and 5) redistribution of Hg inside organisms. Selenium may increase blood Hg, via a “sink” effect, causing a redistribution of mercury away from the brain. A combined approach for mercury poisoning treatment was developed focusing on restoration of selenoprotein function, reduction of oxidative stress and increased mercury elimination.
There has been a paradigm shift in our understanding of the pathophysiology of mercury poisoning and the central role of the interaction between mercury and selenium [Citation1]. Despite this change recommendations for the management of mercury poisoning have remained the same [Citation2–4]. Historically mercury poisoning has been managed by removing the patient from mercury exposure and this has been supplemented by increasing excretion of mercury with thiol-based chelators. However, there are no controlled trials evaluating the clinical benefit of the thiol chelators in mercury exposures. Limitations of thiol-based chelation include ineffectiveness in chronic exposures [Citation5–10] and with the organomercurials such methylmercury (MeHg+), ethylmercury and dimethylmercury [Citation3, Citation11–16].
In the case of another metal, lead poisoning, large studies have shown no neurological benefit after chelation in children with blood lead levels between 20 µg/dL and 44 µg/dL, despite lowering blood lead levels and increasing urinary lead excretion [Citation17]. Additionally chelation in the situation of chronic arsenic poisoning has also been found to be ineffective [Citation2]. These studies bring into question the reliance on increasing urine levels and lowering blood levels as a measure of success of chelation therapy [Citation17].
We propose a reevaluation of the treatment of mercury poisoning, using an evidence-based approach and incorporating the recent advances in understanding of the mechanism of mercury toxicity and the mercury selenium interaction. A search was performed using Medline/PubMed, Toxline, Google Scholar, and Google for published work on treatments of mercury toxicity covering the period 1950 through 2020. Multiple searches were performed using combinations of text words: mercury, mercury toxicity, mercury poisoning, inorganic mercury, organic mercury, methylmercury, dimethylmercury, mercuric chloride, chelation, DMSA, 2,3-dimercaptosuccinic acid, succimer, DMPS, 2,3-dimercaptopropane sulfate, unithiol, selenium, selenide, selenite, diphenyl diselenide, selenoprotein, inorganic selenium, organic selenium, selenomethionine, selenocystine and selenocysteine. The citations were screened for appropriateness and relevancy. Publications that did not evaluate treatment of mercury toxicity or selenium status, or evaluated environmental status (e.g. lake or ocean sediment) were excluded, leaving approximately 350 citations. This initial selection was scrutinized carefully and 178 of the most relevant and representative references were selected for use in this review. Numerous clinical reports provided no dosing or timing of treatments (e.g. “chelation was begun”) or insufficient clinical data for evaluation and were excluded. Please see for a detailed summary and evaluation of references.
This review will focus on four areas:
The mechanisms, limitations and benefits of chelation in mercury poisoning. This is provided because of the past reliance on chelator enhanced elimination as a primary therapy
The role of selenium supplementation in mercury poisoning therapy including: restoration of selenoprotein activity, pro-demeythlating action of selenium, sequestering of mercury via Hg:Se complexes, redistribution of Hg inside organisms under the influence of Se, the inhibitory effects of Se on actions of Hg
How the different forms of mercury are effected by use of chelation elimination and selenium supplementation
Synergy or conflicts between use of selenium, acetylcysteine and the thiol chelators unithiol and succimer
Mechanisms of action and mercury elimination by thiol chelators and acetylcysteine
Thiol-based chelators have been a mainstay of therapy for mercury toxicity for decades [Citation3, Citation4]. However, there are no randomized clinical trials evaluating clinical outcomes after chelation of mercury in humans. The primary role of chelation for mercury poisoning is a proposed reduction of mercury body burden through increased urinary mercury elimination [Citation3]. While it makes intuitive sense that increasing elimination and therefore reducing the mercury body load should reduce the effect of the injury, the lack of improvement in many cases suggests that this simple approach is insufficient.
Much of the early work on thiol-based chelators focused on the use of dimercaprol. However, dimercaprol is contraindicated in organic mercury exposures and may increase brain mercury [Citation14, Citation18, Citation19]. Additional disadvantages of dimercaprol included its intramuscular route, low therapeutic index and toxic effects. Presently the water-soluble oral derivatives of dimercaprol: unithiol (2,3-dimercaptopropane-1-sulfonate; DMPS) and succimer (2,3-dimercaptosuccinic acid; DMSA) [Citation2, Citation3, Citation20] are now preferred.
The primary effects of the water-soluble thiol chelators are: 1) to form a complex with mercury in the proximal tubular cells of the kidney which is then excreted to the tubular lumen via the multidrug resistance protein 2 (MRP2) (and possibly other members of this ATP-binding cassette (ABC) transporter superfamily) and 2) to complex with mercury in the blood (unithiol) and increase clearance via glomerular filtration [Citation20, Citation21] (Figure1). Unithiol and succimer are not able to cross the blood brain barrier or penetrate other target tissues (brain, muscle, thyroid, etc). These other tissues lack a necessary membrane transporter to allow unithiol or succimer cellular penetration, such as the OAT1 in the renal tubules.
A third potential chelator for mercury is acetylcysteine [Citation4, Citation22–35]. As presently understood, the primary role of acetylcysteine appears to be increased mercury excretion via complexation and renal elimination, similar to the thiol chelators (). However additional benefits beyond simple increased urinary Hg excretion have been documented, including: 1) increased demethylation of MeHg, 2) reduced oxidative stress via increased glutathione production, 3) reduced cellular apoptosis, and 4) increased efflux of Methylmercury (MeHg) from the brain may also be important [Citation22, Citation23, Citation27, Citation28, Citation31, Citation33, Citation36–38].
Figure 1. Renal mechanisms for mercury clearance.
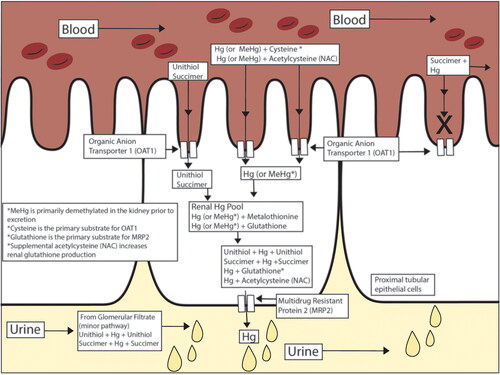
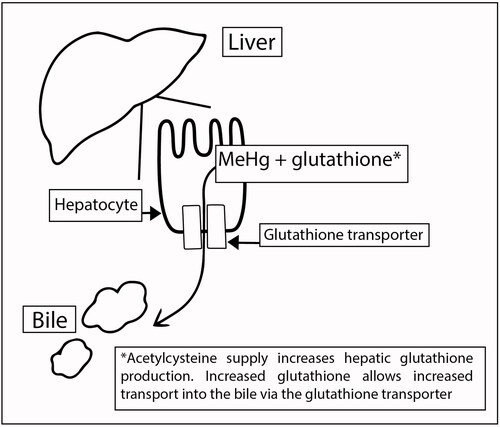
Conjugates of acetylcysteine and MeHg+ or inorganic mercury (Hg2+) form in the blood and act as substrates for the Organic Anion Transporter (OAT1) in the basolateral membrane of the proximal tubular epithelial cells, allowing increased renal uptake of Hg [Citation29, Citation30, Citation39]. After renal uptake, the acetylcysteine-Hg complex is excreted to the tubular lumen by the multidrug resistance protein 2 (MRP2) and possibly other members of this ABC transporter superfamily [Citation33] (). This accelerates the normal pathway of renal elimination of the Hg-cysteine complex as a substrate for OAT1 and the Hg-glutathione complex as a substrate for MRP2 [Citation20, Citation40] (). Additionally: 1) portions of acetylcysteine-MeHg conjugates formed in the blood may be eliminated by the liver into the bile via the glutathione transporter similar to normal MeHg elimination pathway (), and 2) acetylcysteine-Hg conjugates formed in the blood may be eliminated via glomerular filtration similar to acetylcysteine clearance in the urine and the unithiol-Hg complex.
Figure 2. Hepatic mercury elimination.
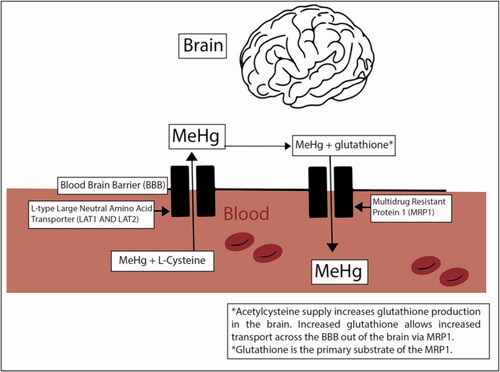
Acetylcysteine is able to cross the blood brain barrier and is available to all tissues. Increased neuronal glutathione production from acetylcysteine can increase efflux of MeHg+ from the brain via the glutathione + MeHg complex using the multidrug resistance protein and reduce brain MeHg+ concentrations [Citation41, Citation42] (). A number of studies involving in vitro neurons, astrocytes, neuroblastoma cells and myogenic cells incubated with MeHg+ or Hg+2 and treated with acetylcysteine showed reduced oxidation injury, increased cell viability and blocked Hg-induced reduction of DNA synthesis [Citation36, Citation37, Citation43, Citation44].
Figure 3. Transport of Methylmercury across the blood brain barrier.
Chelation – limitations, blood and urine mercury changes may not equate with clinical improvement
An important limitation of unithiol and succimer is inability to directly remove Hg from target organs such as the brain and liver, due to lack of tissue penetration. While acetylcysteine does assist increased efflux of MeHg from the brain, no chelator appears to effect inorganic Hg brain content directly. A second important limitation of the thiol chelators Unithiol and Succimer is after chronic mercury exposure, decreases in blood Hg concentration or increases in urine Hg concentration do not necessarily equate to improved clinical outcome [Citation12, Citation13, Citation16, Citation45–49]. This is likely because changes in blood and urine mercury levels do not address cellular selenium deficit, selenoprotein inhibition and subsequent cellular mitochondrial injury [Citation1].
Clinical reports using chelation therapies for mercury poisoning have shown significant increases in urine mercury concentrations, but have produced conflicting reports of patient outcomes including: renal, cardiovascular and/or neurological improvement [Citation50–55], prevention of toxicity [Citation56–58], no effect (lack of improvement) [Citation6, Citation7, Citation49, Citation56, Citation59–65], and continued patient deterioration [Citation12, Citation13, Citation16, Citation46–49, Citation66, Citation67]. The inconsistent results seen following chelation draws into question the reliance on changes in blood and urine Hg concentrations when assessing the effectiveness of therapies. Though there have been patients who experience significant clinical improvement following chelation [Citation68–72] (even in patients without elevated blood and urine Hg levels [Citation50, Citation52]), there are also numerous cases reporting a lack of improvement and even continued deterioration [Citation12, Citation13, Citation16, Citation46–49, Citation66, Citation67].
In six reports [Citation13, Citation51, Citation61, Citation73–75] describing unithiol or succimer therapy in patients with either elemental mercury, mercurous chloride or thiomersal [ethymercury] exposure (18 patients in total with daily urinary mercury concentrations), urine mercury concentrations rose dramatically during the first day but returned to near pre-chelation concentrations over the following consecutive days, despite continued therapy [Citation13, Citation51, Citation61, Citation73–75]. Pfab et al. [Citation13] using an alternating oral and IV unithiol (days 1–16 and 23–29) and oral succimer (days 17–22 and 33–70) reported more mercury (as thiomersal) was excreted in the first 3 days than the entire remaining 70 days of chelation [Citation13]. In this case brain levels continued to rise for 18 days with continued patient deterioration, despite ongoing chelation and falling urine mercury levels [Citation13]. This supports the mechanistic understanding that the unithiol and succimer appear to have only limited effect on target organ mercury beyond the kidney.
Likely because of the noted decrease in urine mercury output with continuous dosing, a number of clinicians have employed repeated “pulse” dosing with chelation free periods [Citation51, Citation74–76]. Four reports describe the use of a repeated “pulse” dosing of unithiol:
chronic mercury vapor exposure treated with 30 mg/kg/day (3000 mg/day) for five days repeated three times over 9 weeks [Citation51]
chronic mercury vapor treated with 600 mg/day for 14 days with a 38 days break and then 800 mg/day for 20 days [Citation74],
chronic occupational mercurous chloride exposure treated with 400 mg/day for 8 days followed by 5 days free of unithiol, then unithiol 400 mg/day orally for 7 days, followed by 5 days free of unithiol, then 6 days of unithiol 400 mg/day orally [Citation75],
chronic mercury exposure in a 10 month old treated with unithiol 20mg/kg/day IV for 4 days followed by 4 cycles of 20 mg/kg/day unithiol IV for 4 “cycles” of 2 days each [Citation76].
In all four cases the second, third and fourth unithiol treatments resulted in urine mercury concentrations less than 10% of the first unithiol course [Citation51, Citation74–76]. Additionally, the majority (>75%) of urine mercury excretion occurred in the first 48 h of the first treatment, supporting the animal data that the primary effect of the thiol chelators is reduction of renal stores of mercury [Citation51, Citation74, Citation75, Citation77]. In chronic mercury poisoning or post distribution phase, unithiol and succimer appear to have only limited effect on target organ mercury beyond the kidney [Citation78].
Several other features may effect clinical outcome greater than simple comparison of urine or blood concentrations. The forms of mercury (elemental mercury and the organic and inorganic salts) have critical differences in target organ, toxicokinetics, clinical presentation, and potency [Citation79, Citation80]. Chronicity, allowing distribution into target organs, especially the brain, is important [Citation45]. More importantly, a majority of clinical reports and animal studies do not address selenium status or the mercury/selenium interaction. This failure to include assessment of the primary mechanism of toxicity of mercury in the majority of reports may explain some of the conflicting differences in outcome, when using only mercury concentrations as a measure
Mercury kinetics after acute exposure and effect of chelation
The half-lives of organic, inorganic and metallic mercury vary significantly, as do the clearance from target organs such as the brain [Citation81, Citation82]. The kinetics of the forms of mercury in humans are poorly understood and are quite different depending on the type of Hg. Several human reports in both inorganic and organic Hg suggest a 2-compartment model with an initial distribution phase, supported by significant initial drops in blood Hg, despite in some cases no treatment or total anuria with no urinary mercury elimination [Citation58, Citation69, Citation72]. We suggest caution in acute exposures when attributing early drops in blood Hg to effects of chelation, without understanding the distribution phase of that form of Hg.
Chelation: choice of chelator depends on the form of mercury
Choice of a chelator with organic mercury – acetylcysteine
For methylmercury (Hg+1) and dimethylmercury, acetylcysteine appears to be a better choice of chelator [Citation22, Citation24, Citation26, Citation27, Citation32–34, Citation68, Citation83]. Acetylcysteine increases urinary clearance, reduces target organ mercury levels, including increased efflux from the brain, as well as shows improvement in organ function (brain, liver and kidney) [Citation22, Citation26, Citation27, Citation32, Citation33, Citation68, Citation83]. The remediation of neurotoxicity is likely due, in part, to the protective increased glutathione production in the brain, a unique feature of acetylcysteine [Citation22, Citation31, Citation36, Citation44, Citation83].
Koh et al., compared acetylcysteine, unithiol, succimer, and l-cysteine as substrates for the OAT1 transporter in an in-vitro model of MeHg exposure [Citation30]. Acetylcysteine, and unithiol were equally good substrates for OAT1, and were significantly better substrates than succimer or l-cycteine [Citation30].
With organic mercuries (methylmercury, dimethylmercury, and ethylmercury) the thiol chelators (unithiol and succimer) have shown increases in urinary mercury and in some cases decreases in blood mercury. Unfortunately, even with these mercury concentration changes there has been limited or no clinical improvement in the patients, with numerous cases reporting continued patient deterioration, in some cases to permanent disability or death [Citation12, Citation13, Citation16, Citation45, Citation59, Citation66]. This likely reflects in vivo demethylation of the organomercuries in the brain [Citation81, Citation82]. The inability of the thiol chelators to cross the blood brain barrier and access demethylated inorganic mercury (Hg+2) is likely responsible for its poor remediation of neurotoxicity associated with the organomercuries [Citation15].
Ethylmercury has a shorter half-life and greater difficulty crossing the blood brain barrier than methylmercury [Citation84]. Unithiol and succimer have shown limited or no effect on neurotoxicity from ethylmercury when used in humans [Citation13, Citation50, Citation69, Citation85]. Similar to methylmercury this likely reflects dealkylation of ethylmercury to inorganic mercury (Hg+2) in the brain and the inability of unithiol of succimer to cross the blood brain barrier [Citation86, Citation87]. Acetylcysteine has not been evaluated with ethylmercury in humans, but the additional role of increased glutathione production in the brain supports the choice of acetylcysteine [Citation88].
Choice of a chelator with elemental mercury and mercury vapor
Elemental mercury (injection) and mercury vapor are absorbed in the un-ionized state (Hg0). This allows for passive diffusion into organs and across the blood brain barrier. Hg0 is then oxidized to the toxic form Hg2+ after tissue penetration, including in the CNS, RBC, and kidney [Citation9]. The hypertension and tachycardia associated with acrodynia are likely due to intracellular calcium dyshomeostasis (selenoprotein control of endoplasmic reticulum function with increased intracellular release of calcium) in the adrenal glands [Citation1].
In elemental mercury and mercury vapor poisoning the thiol chelators (unithiol and succimer) have shown 1) increases in urinary mercury, 2) decreases in blood mercury, and 3) reductions of kidney and total body Hg [Citation9, Citation51, Citation77]. Neither the thiol chelators nor acetylcysteine show any effect on brain mercury concentrations after mercury vapor exposure [Citation8, Citation9, Citation15, Citation77]. This may partially explain the varying responses in the clinical reports after vapor exposure. Acute asymptomatic cases show increased urinary mercury and reduced blood levels and remain asymptomatic [Citation70, Citation73]. However, chronic exposure after onset of neurotoxicity is generally manifested by prolonged recovery with or without permanent neurologic injury despite increased urine Hg and decreased blood Hg [Citation46, Citation51, Citation56, Citation60, Citation61, Citation76, Citation89]. Additionally, in acute high-concentration mercury vapor cases unithiol or succimer appear to have limited effect on pulmonary injury [Citation60, Citation88]. Similar to cases of mercury vapor poisoning, chelation appears to be inadequate to manage the large multi-gram doses associated with elemental mercury injection [Citation57, Citation74, Citation90].
Acetylcysteine does not appear to decrease tissue mercury after vapor exposure but reduces organ injury in animal studies, likely through a separate mechanism such as increased intracellular glutathione [Citation9, Citation38, Citation43].
Choice of a chelator with inorganic mercury salts
In animal studies of mercuric chloride injection unithiol decreased mercury content in all organs, while succimer decreased only kidney and blood concentrations, if therapy was begun within a day of mercury exposure [Citation8, Citation11, Citation19]. However, if there was a delay in chelator administration, therapy was less effective and brain mercury was not improved [Citation10, Citation91, Citation92]. This again highlights the lack of tissue penetration of unithiol or succimer. Conversely, in mice, prolonged use of unithiol or succimer (4 weeks) increased Hg content of spinal motor neurons 2-fold, likely a consequence of increased blood levels of mercury redistributed from non-neuronal tissues by the chelators [Citation93]. Acetylcysteine after HgCl2 has also produced conflicting results, with 1) no effect on mercury organ content [Citation27], or 2) reduced kidney and liver mercury content along with restoration of organ function [Citation23, Citation28]. In clinical reports of ingestion and dermal exposure to inorganic mercury unithiol and succimer increased urine mercury and decreased blood hg significantly (See limitations, Blood and Urine mercury changes above) [Citation51, Citation75, Citation94, Citation95].
For both inorganic mercury and elemental mercury/vapor the initial use of a thiol chelator for 3 to 4 days (see Blood and Urine mercury changes above), to clear renal Hg may be appropriate. However, after 3–4 day the switch to acetylcysteine for continued chelation, reduced organ injury, and synergy with Se is suggested.
Dose and timing of chelators
The effect of timing, route of administration (bioavailability) and dose of unithiol on the elimination of mercury is complex and requires explanation. The water-soluble thiol chelators lack the ability to penetrate tissues and therefore are most effective when they are able to access mercury in the blood and kidney prior to tissue distribution [Citation15, Citation20, Citation21, Citation77, Citation92]. In animal studies, if given within 5 days (prior to mercury distribution) unithiol was effective at reducing total body mercury and all organ mercury content. This is likely due to access to mercury still in the blood prior to distribution [Citation77, Citation92]. However if administration is delayed beyond 5 days unithiol is only effective in reducing blood and kidney mercury with no significant effect on mercury content of other important target organs such as the brain and liver [Citation15, Citation77, Citation92].
Data obtained from animal studies suggest that the primary effect of the thiol chelators is reduction of renal stores of mercury [Citation77, Citation78]. In two human cases of chronic elemental mercury exposure, use of unithiol 600 mg/day orally resulted in peak urine mercury excretion of 2000 µg/L [Citation90], while quadrupling the dose resulted in peak urine mercury concentrations of only 1500 µg/L [Citation57]. In unithiol chelation studies involving both animals and humans, urine mercury decreases rapidly after 2–4 days, with urine mercury levels returning to near pre-chelation levels despite continued therapy even when high doses are used (up to 200 mg/kg IV) [Citation10, Citation13, Citation15, Citation51, Citation57, Citation73–75, Citation77].
Oral bioavailability of unithiol in humans is only approximately 40% [Citation96]. Therefore, some authors suggest that current oral dosing regimens for unithiol are inadequate and should be increased [Citation51, Citation96]. Though the relationship between urine mercury concentration and increasing oral unithiol doses is linear in the case of early presenting, acute mercury toxicity, this is not the true in the setting of chronic poisoning and cases of delayed presentation, when tissue distribution of mercury is complete [Citation15, Citation77, Citation92]. In these cases, there is no apparent benefit to increased unithiol doses.
Increasing the dose of acetylcysteine increases MeHg+ excretion but not Hg+2 in a proportional manner [Citation34]. In rats, given a MeHg dose of 0.1 µmol/kg (21.4 µg/kg), increasing the dose of acetylcysteine from 0.25 mmol/kg (40.5 mg/kg) to 1.5 mmol/kg (244.8 mg/kg) showed a near linear increase in mercury elimination, from 2% of total dose eliminated in 2 h to 10% of total dose eliminated in 2 h, respectively, compared with 0.1% elimination in controls (MeHg+ but no acetylcysteine) [Citation34].
Selenium
Selenium supplementation
Early studies in cats and quail suggested selenium supplementation might provide a protective effect from mercury toxicity [Citation97, Citation98]. Similar “protective” effects have been reported in additional animal models and humans [Citation99–104]. However, more recent studies on the pathophysiology of mercury, suggest selenium supplementation may not only be protective, but also corrective of a mercury-induced selenium deficiency state [Citation1, Citation105]. There is convincing evidence that the primary target of mercury is inhibition of the selenoproteins in the thioredoxin system (thioredoxin reductase) and glutathione-glutaredoxin system (glutathione peroxidase), along with secondary involvement of selenoproteins P, K, T, and W [Citation1, Citation105–112]. Mercury binds to the selenium moiety of the selenoprotiens with high affinity, which unless removed permanently inhibits their activity. The loss of selenoprotein activity causes disruption of the cellular redox environment, lipid and protein peroxidation, disruption of intracellular calcium homeostasis, mitochondrial injury/loss and an intracellular selenium deficiency [Citation1].
The role of selenium supplementation in mercury poisoning is complex and includes 1) restoration of selenoprotein activity, 2) protection against mitochondrial injury and DNA damage, 3) pro-demethylating action of selenium, 4) sequestering of mercury via Hg:Se complexes, 5) redistribution of Hg inside organisms under the influence of Se, and 6) the inhibitory effects of Se on actions of Hg [Citation99, Citation113–123]. However, similar to the limitations of increased urinary elimination of Hg by the thiol chelators, the amelioration of mercury toxicity from simple resupply of selenium has important limitations [Citation105, Citation119, Citation124].
Numerous studies of simultaneous administration of selenium and mercury over extended periods showed beneficial effects as evidenced by reduced mercury tissue concentrations, reduced mercury whole body retention, improved thioredoxin reductase activity, improved glutathione peroxidase activity, reduced oxidative stress, reduced DNA damage, mitochondrial rescue, improved muscle strength and body weight gain [Citation100, Citation102, Citation114, Citation117, Citation124–129]. Study methods have used Mercury vapor (Hg0) with oral selenium for 4 weeks in rats [Citation126] oral MeHg+ with diphenyl diselenide injection in mice [Citation117], oral mercury (MeHg+ and Hg+2) with oral selenite for 14 days in mice [Citation128], oral MeHg+ with oral selenate for 100 days in rats [Citation126], oral MeHg+ with oral selenite for 16 months in rats [Citation130], oral MeHg+ with injection of diphenyl diselenide in mice for 21 days [Citation129], and oral MeHg+ with oral selenite for 19 weeks in rats [Citation100]. While these studies are important in understanding the mechanism and interaction of selenium and mercury, they show primarily evidence of adequate or supratheraputic selenium status during ongoing mercury poisoning. The studies do not address what benefit selenium supplementation might provide if begun after mercury toxicity has already occurred, which is the question often faced in human events. However, a number of studies looking at effects of selenium supplementation initiated after mercury exposure have reported positive effects in both organic and inorganic mercury [Citation22–24, Citation48, Citation81, Citation83, Citation131–135].
Selenium acetylcysteine synergy
Several studies have reported improved outcomes from the combined use of selenium supplementation and acetylcysteine [Citation22–24, Citation81].
Use of selenium and acetylcysteine with inorganic mercury poisoning – mercuric chloride (Hg + 2)
Joshi et al. [Citation23] looked at selenium administration as selenite with and without acetylcysteine given after mercury exposure had ceased. Using HgCl2 (2.83 mg/kg i.p. × 1) in rats and waiting 24 h to begin selenium (500 µg/kg/day times 3 days) and/or acetylcysteine (0.6 mg/kg/day times 3 days), Joshi et al. [Citation23] showed reduced lipid peroxidation, improved oxidation status, restored kidney function, improved liver function and reduced measures of liver injury (bilirubin, cholesterol, triglycerides, AST, ALT, and LDH) when compared with HgCl2 poisoned rats receiving no treatment. Selenium alone was more effective than acetylcysteine alone, but selenium plus acetylcysteine together restored the rat (hepatic function, renal function and hisptopathoogical exam of liver and kidney post mortem) to within 80% of control (no HgCl2) in four days, at the time of sacrifice [Citation23]. Additionally, selenium and acetylcysteine administered together reduced kidney and liver mercury concentrations by 70% and 80%, respectively, in four days, at the time of sacrifice compared with HgCl2 poisoned rats receiving no treatment [Citation23].
Use of selenium and acetylcysteine with organic mercury poisoning – methylmercury (MeHg+) and dimethylmercury
Similar results (improved oxidation status, improved organ function and reduced injury) were found in a methylmercury rat model (MeHg+ 1.5 mg/kg/day orally for 21 days), with a 24 h delay followed by selenium (500 µg/kg/day orally for 5 days) and/or acetylcysteine (0.6 mg/kg/day i.p. for 5 days) [Citation22]. Selenium alone was more effective than acetylcysteine alone, but selenium plus acetylcysteine together restored the rat (reduced brain lipid peroxidation, increased brain acetyl cholinesterase and increased brain, liver and kidney glutathione) to within 80% of control (no MeHg+) in 7 days, at the time of sacrifice [Citation22]. Additionally, selenium and acetylcysteine administered together reduced brain, kidney and liver mercury concentrations by 87%, 88% and 86%, respectively, in 7 days, at the time of sacrifice compared with MeHg+ poisoned rats receiving no treatment [Citation22].
In a model using dimethylmercury, perhaps the most neurotoxic form of mercury, selenium with acetylcysteine and zinc showed promising results. Joshi et al. [Citation24] used three groups 1) controls (no mercury and olive oil as vehicle for 5 days), 2) dimethylmercury 1.5 mg/kg/day orally for 21 days, with a 24 h delay, and olive oil as vehicle for 5 days, and 3) dimethylmercury 1.5 mg/kg/day orally for 21 days with a 24 h delay followed by 5 days of selenium 0.5 mg/kg p.o. plus acetylcysteine 624 mg/kg i.p. plus zinc 2 mM/kg p.o. The treatment group showed significant reduction of brain, kidney and liver mercury concentrations by 46%, 64%, and 43%, respectively, compared to Hg poisoned rats with no treatment group [Citation24]. Additionally, treated rats show significant restoration of organ function including brain (acetylcholinesterase activity and adenosine tryphosphate), kidney (adenosine triphosphate, creatinine, BUN and urea) and liver (bilirubin, serum triglicerides, gamma glutamyl tranpeptidase, and adenosine triphosphate), compared with no treatment group [Citation24]. Histopathological examination of the three groups showed restoration of the treatment group near to controls (no Hg and no treatment) and compared with significant injury in the Hg only group [Citation24]. In the treatment group brain there were well formed nerve filaments, recovered glial cells and the number of neurons and nerve fibers were restored, compared with reduction of nerve fibers, neuronal degeneration and fusion of nerve bundles in the Hg no treatment group [Citation24]. Similar restoration of histopathological structure in the kidney and liver to near controls was seen in the treatment group [Citation24].
Use of selenium and acetylcysteine with mercury vapor
A 15-year-old boy with mercury vapor exposure presented with a 2-month progression of neurologic and cardiovascular symptoms, including hypertension, tachycardia, tremor, insomnia, muscle pain, diaphoresis, altered gait, weight loss and a 1-month removal from mercury exposure, with a blood mercury concentration of 30 μg/L [Citation48]. He was treated with oral Se 500 mcg/day (as sodium selenite) and oral acetylcysteine 50 mg/kg/day associated with significant clinical improvement including weight gain (50 pounds, 22.7 kg over 3 months), increased muscle strength, correction of hypertension, tachycardia, and tremor. Within 90 days there was full return to age appropriate norms for weight, physical and academic performance and resolution of neurological (tremor, gait) and cardiovascular (heart rate, blood pressure) symptoms [Citation48].
Selenium alone – methylmercury
In a rat model of chronic methylmercury exposure until evidence of toxicity (4 mg/kg MeHg+ every other day via oral gavage for 4 weeks), the effects of selenium supplementation was evaluated [Citation134, Citation135]. In comparing Se (2.74 mg/kg as sodium selenite via oral gavage every other day after MeHg+ exposure had ceased, equivalent to a 1:1 Hg:Se molar ratio) to no Se controls (saline via gavage), the Se treated rats showed increased weight gain, reduced oxidative damage and reduced Hg in the brain, liver and blood, compared with controls (no Se) [Citation134, Citation135]. In these animals Se supplementation increased selenoprotein P, with 73% of Hg bound to selenoprotein P, suggesting this may be the mechanism for sequestration of Hg from target organs and transport for elimination [Citation135]. In a rat model of MeHg, three groups were compared 1) control no Hg, no Se, 2) MeHg (4 mg/kg/day for 4 weeks) and 3) MeHg (4 mg/kg/day for 4 weeks) with Se as selenite (1.5 mg/kg/day), Se treatment ameliorated evidence of apoptosis and autophagic dysfunction in the pituitary and protected histopathological structures [Citation136]. Additional in vitro evidence showed selenocysteine increased hepatic uptake of MeHg+ as a Se-MeHg complex, and reduced cytotoxicity of the MeHg [Citation137].
In a chronic methylmercury rat model (MeHg+ 4 mg/Kg p.o. every other day for 28 days) the addition of selenium (Se 2.74 mg/kg p.o., equimolar dose 1:1 to MeHg) on day 29 for 90 days, showed restoration of gut flora, increased total mercury in feces and decreased the portion of MeHg+ in feces, supporting increased demethylation [Citation138]. This was a small study of only 5 rats per group but this study suggests that Se promoted the decomposition and excretion of methylmercury, which can be partially ascribed to the modulation of gut flora [Citation138].
Clinical studies of selenium in mercury poisoning
Residents (n = 103) of a mercury mining and smelting district in Wanshan China with elevated urine mercury concentrations were either administered selenium 100 µg/day (n = 53) or placebo (n = 50). Selenium supplementation significantly reduced markers of oxidative stress, as measured by urinary malondiadehyde and 8-hydroxy-2-deoxyguanosine levels [Citation132]. Additionally selenium significantly increased urine mercury concentrations compared with controls: from a mean of 18 µg/L to 50 µg/L vs no change, respectively [Citation132]. In a similar group of 5 residents with elevated urine Hg, Se supplementation increased Urine Hg from mean 173 µg/L (day 0) to 1018 µg/L (day 90). Ninety-five percent of Hg excreted was inorganic Hg. Urine Se did not change with supplementation [Citation131]. The increase in urine mercury excretion did not begin for 15 to 30 days, suggesting redistribution of tissue mercury, rather than a direct renal effect [Citation131, Citation132].
Draz et al. [Citation133] compared 36 lamp factory workers in Egypt with ongoing occupational exposure to mercury, lead and cadmium to 20 non-exposed age matched controls [Citation133]. Daily supplementation with selenium 100 µg/day for 2 months and vitamin E 100 mg daily for 2 months significantly (p < 0.05) reduced blood mercury concentrations in exposed workers (22%) and returned markers of oxidative stress (serum malondiadehyde) to those of non-exposed controls [Citation133].
There is evidence that adequate or supratherapeutic selenium status provides benefits in the presence of elevated blood mercury or total body mercury concentrations [Citation139–142]. Li et al. [Citation143] reported on 4 groups from 4 geographic areas (3 groups from a mercury mining region and 1 from a non-mining region) in Wanshan China. In the 3 mining areas blood total Hg and blood MeHg+ were significantly elevated (means 12.5 to 14.7 µg/L and 6.64 to 7.93 µg/L, respectively) compared with the non-mining region (means 5.5 µg/L and 3.83 µg/L). However serum selenium concentrations were also significantly elevated in the 3 mining region groups vs the non-mining region group (219 to 274 µg/L vs 157 µg/L), with all groups showing an Se:Hg molar ratio of >10, suggesting a protective effect [Citation143].
Lemire et al. [Citation139] reported on a group from the Amazon basin with high mercury contamination (Median blood mercury concentration 42.5 µg/L), presumably from dietary sources. While elevated blood mercury concentrations showed a negative association with motor function tests, residents with elevated plasma selenium concentrations (median 135 µg/L, also from dietary sources) showed amelioration of these effects and positive association with improved motor functions.
Nakamura et al. [Citation140] looked at a group in Taiji Japan with a history of whale meat consumption and measured blood mercury, hair mercury and blood selenium. Despite having chronic mercury exposure (hair mercury >50 µg/g) they did not detect neurologic impairment expected from chronic mercury exposure. They attributed the lack of neurologic impairment to the near equimolar mercury/selenium balance found in these residents [Citation140].
Dose of selenium
In humans with mercury poisoning, 100 µg/day for 90–120 days (as selenomethoinine) and 500 µg/day for 6 months (as selenite) have been used successfully [Citation48, Citation131–133]. While the upper level of safety with selenium supplementation is not defined, selenium supplementation in humans of up to 800 µg/day for 16 weeks (as selenite and selenomethionine) showed no evidence of toxicity [Citation141]. In humans chronic ingestion of sodium selenite 40, 800 µg/day for a median of 29 days produced significant selenosis and mean serum selenium levels of 761 µg/L [Citation142].
In mercury toxicity studies using rats, larger doses of selenium for shorter periods of time have been used with positive effects (500 µg/kg/day for 4 to 7 days and 865 µg/kg/day for 21 days) [Citation22, Citation23, Citation83, Citation119]. However, larger doses (32 mg/kg as diphenyl diselenide) appeared to increase lethality [Citation144]. Conversely, in an animal model of selenium toxicity (40 ppm Se in daily feed, total quantity of Se/day not measured), the addition of Hg+2 (500 ppm, 1000 ppm as HgCl), significantly reduced the toxicity of the Se, as measured by weight gain and percent mortality at 2 and 3 weeks [Citation145].
In the presence of Hg2+ 5 µmol, selenium 2 µmol was more effective than the thiol chelators dimercaptol, unithiol and succimer in restoring thioredoxin reductase activity [Citation125]. However, in the presence of Hg2+ 10 µmol, it required selenium 8 µmol for significant rescue. For comparison 1 µmol of Hg2+ in cortical cell cultures (containing both neurons and glial cells) caused 40% cell death and 5 µmol Hg+2 caused 100% cell death [Citation146]. For comparison, in one human case peak clinical effects occurred with a CSF of thiomersal (ethylmercury) of 0.11 µmol (25 µg/L) [Citation13]. Normal human reference plasma selenium concentrations range from 50 to 160 µg/L [Citation147]. This equates to solutions containing selenite 0.5–2.0 µmol/L [Citation143]. However, healthy populations have been recorded with higher serum selenium in the range of 219 to 274 µg/L [Citation148, Citation149].
Effects on mercury kinetics after selenium supplementation
Selenium demethylation of organic mercuries and sequestration of mercury
Selenium supplementation may increase demethylation of organic mercuries, change mercury distribution and increase sequestration via binding to colloidal insoluble Hg:Se complexes. Inorganic mercury (Hg2+) does not readily cross the blood brain barrier, while both mercury vapor (Hg0) and MeHg+ more readily cross the blood brain barrier. The mechanism of transfer across the blood brain barrier might be passive infusion (Hg0) or as a substrate for the L-type large neutral amino acid transporter (MeHg+ and likely ethylmercury) [Citation8, Citation80, Citation128, Citation150–152] (). Once in the brain, portions of both Hg0 (oxidation) and MeHg+ (dealkylation) are converted to Hg2+ and are therefore trapped in the brain [Citation8, Citation81, Citation82, Citation153, Citation154].
In monkeys, a chronic diet-source MeHg+ model demonstrated that the estimated half-life of MeHg+ in the brain was 37–45 days, but increased to 230–300 days for the demethylated inorganic Hg2+ [Citation81]. Brain inorganic Hg was formed by demethylation in the brain, not by brain uptake of inorganic Hg demethylated elsewhere in the body [Citation81].
Selenium supplementation and selenoprotein activity is likely responsible for in vivo demethylation of MeHg+ and conversion to inorganic Hg2+ in tissues, including the brain [Citation101, Citation103, Citation138, Citation153, Citation155]. In comparing chronic exposure in mice (30 days), rats (18 weeks) and post-natal rat pups (10 days), both mercury exposure groups (mercury vs mercury plus selenium) showed increases in brain mercury, but the selenium supplementation groups did not display the clinical and pathological evidence of mercury toxicity [Citation100, Citation102, Citation156]. In a rat model of oral MeHg+, (MeHg+ 40 mg/L in freely available water for 21 days), daily injection of Se (as sodium selenite 865 µg/kg/day i.p. for 21 days) effectively eliminated deposition of mercury in the brain [Citation119].
The supplemental selenium may produce this protective effect by binding with the available mercury, producing an inert colloidal mercury:selenium complex [Citation101, Citation119, Citation120, Citation127, Citation135, Citation137, Citation155, Citation157–159]. Support for this theory comes from autopsy studies in occupationally exposed mercury mine workers in Indrija Slovenia (compared with non-exposed population), who had elevated brain mercury content found 5 to 16 years after retirement [Citation157]. Autopsy results compared three groups: 1) occupationally exposed mercury mining workers, 2) non-occupationally exposed residents from the mining district (Indrija Solvenia), and 3) non-exposed population from a non-mining region of Slovenia [Citation157]. Despite a 20 fold increase in brain Hg (occupationally exposed vs non-occupational exposed local residents) and a 100 fold increase in brain Hg (occupationally exposed vs non-exposed from a non-mining region) all autopsies showed a near 1:1 molar Hg:Se ratio in the brain, thyroid and kidney, suggesting an active biological sequestration via an inert Hg:Se colloidal complex [Citation157].
While it seems intuitive that redistribution away from target organs and increasing mercury elimination are important treatment goals, it may be that complexing with and sequestering already distributed mercury is an additional role of selenium supplementation [Citation119–121].
Effect of selenium supplementation on selenoprotein activity
Selenoproteins are critical endogenous proteins that regulate: 1) the cellular redox environment protecting against protein and lipid peroxidation within the cells (e.g. thioredoxoin reductase, glutathione peroxidase); 2) calcium release from the endoplasmic reticulum (e.g. selenoprotien T, K, W); and 3) selenium delivery to organs and mercury distribution (e.g. selenoprotein P) [Citation1, Citation160].
In in vitro models of Hg+2 poisoning, activity of the selenoproteins thioredoxin reductase and glutathione peroxidase was restored by selenium supplementation (as selenite) [Citation105, Citation107, Citation125]. Selenium supplementation may produce restoration of selenoprotein activity via several pathways: 1) supply of selenium needed during de novo regeneration of the selenoproteins; 2) direct scavenging of the mercury to a colloidal mercury:selenium complex; or 3) stimulation of increased genomic signaling that ultimately leads to enhanced selenoprotein expression [Citation105, Citation107, Citation122, Citation125, Citation161]. Additionally, in a mouse model of MeHg+ exposure, selenium as diphenyl diselenide was able to restore mitochondrial function and increase mitochondrial biogenesis, likely by activating transposition of the nuclear factor E-2-related factor 2 to the nucleus [Citation114, Citation129]. Selenium supplementation may ultimately perform better than the thiol chelators at restoring selenoprotein function, as the thiol chelators lack the ability to penetrate into the CNS. In an in vitro model of rat hepatocytes treated with HgCl2 and MeHg, selenium at 0.5–1 µmol was as effective at restoring thioredoxin reductase activity as unithiol (10 µmol), succimer (10 µmol) or BAL (10 µmol) [Citation125]. However, unlike selenium, unithiol and succimer lack the ability to penetrate the CNS, so restoration of selenoprotein activity in the brain following administration of the thiol chelators would not be expected [Citation8]. In an in vivo model, unithiol showed no effects on the activity of the CNS selenoprotein, neuronal gluthathione peroxidase [Citation162]. In an in vitro model, Se at 50 nmol was able to restore Selenoprotein W production (responsible for cellular calcium homeostasis) after incubation with 1.4 µmol MeHg [Citation163].
Selenium alone was less effective in restoring function of the CNS selenoproteins thioredoxin reductase 1 and 2 inhibited by MeHg+, compared with inhibition by HgCl2 [Citation105, Citation115, Citation123]. MeHg+ may need to be demethylated prior to direct benefits from selenium, however selenite in the presence of glutathione increased demethylation of MeHg+ [Citation22, Citation103, Citation117, Citation164].
Effect of selenium on mercury blood concentrations – redistribution away from target organs
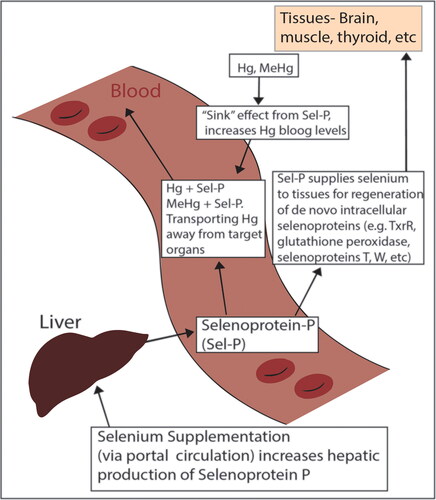
Selenium supplementation may initially result in an increase in blood mercury concentrations [Citation48, Citation99, Citation123, Citation131, Citation132, Citation135]. This is a result of increased selenoprotein P production in the liver [Citation135, Citation165]. Selenoprotein P is a selenium-rich (10 selenocysteines) serum selenoprotein that acts partially as a “sink” for tissue mercury and a transport of Hg:Se complexes away from target critical organs [Citation99, Citation123, Citation134, Citation135, Citation160, Citation165] (see ). In rats poisoned until neurological symptoms appeared (4 weeks of 2 mg/kg/day MeHg+ orally) selenium supplementation begun after Hg exposure ceased (150 µg/kg/day orally as selenite for 90 days) showed a 30% increase in serum mercury, suggesting a sink effect of mercury from tissues [Citation99]. In a similar MeHg+ model of rats poisoned until evidence of neurotoxicity, Se supplementation begun after Hg exposure ceased, significantly increased selenoprotein P levels in the blood, allowing for increased distribution of mercury away from target organs like the brain [Citation135]. In a third methylmercury rat study poisoned until neurological symptoms occurred, Hg in serum increased 240% (50 µg/l to 120 µg/l) at 30 days after selenium supplementation and 300% (50 µg/l to 150 µg/l) at 90 days, again suggesting a significant tissue sink effect [Citation123]. The mercury in serum was bound to high molecular weight proteins in the 170 KDa range, likely selenoprotein P [Citation123, Citation135]. A similar increase in blood mercury, with associated clinical improvement, was seen in one human case after selenium and acetylcysteine were initiated (500 µg/day and 50 mg/kg/day orally, respectively) [Citation48]. Selenium status and the mercury:selenium molar ratio is a more important indicator of patient status than blood mercury concentrations [Citation48, Citation100, Citation124, Citation132, Citation166].
Figure 4. Actions of Selenoprotein-P in mercury poisoning.
Effect of thiol chelators vs acetylcysteine on selenoprotein activity and interaction with selenium
There is a potential problem when considering use of unithiol or succimer together with selenium supplementation. One of the initial effects of selenium supplementation is a redistribution of mercury secondary to increased serum selenoprotein P. In the early phases (days to weeks), selenium supplementation produces increased liver mercury and decreased kidney mercury concentrations [Citation99, Citation156, Citation167–169]. Over time this effect tends to revert to normal distribution patterns [Citation1]. This early redistribution may pose a problem when selenium supplementation is used simultaneously with the thiol chelators, as the primary source of excreted mercury for the thiol chelators is kidney stores of mercury [Citation168, Citation169]. In rats and mice with a single acute mercury injection, simultaneous administration of selenium supplementation (either as diphenyl diselenide or sodium selenite) and unithiol or succimer, showed lower urine mercury concentrations, compared with succimer or unithiol alone [Citation168, Citation169]. Additionally, the thiol chelators may have an effect on tissue disposition and elimination of selenium: reducing serum selenium concentrations, reducing selenium availability for cells and increasing urine selenium excretion [Citation170–172]. Conversely, simultaneous use of selenium with acetylcysteine appears to be synergistic, with increase mercury elimination, improved organ function and reduced mitochondrial injury [Citation22, Citation23, Citation83, Citation119]. Additionally acetylcysteine promotes efflux of Hg from the brain via increased glutathione-MRP1 transport [Citation41].
Failure of selenium supplementation
Comparison of outcomes from selenium supplementation studies can be difficult because of differences in doses used (mercury and selenium), model used (e.g. mouse vs rat), duration of the study and moieties used (e.g. MeHg+ vs Hg+2, selenite vs diphenyl diselenide).
In an in vitro study of seal leukocytes, co-incubation of selenite (7.5 µmol) or selenomethionine (7.5 µmol) with MeHg+ (0.75 µmol) provided no protection against mercury-induced inhibition of cell proliferation [Citation173]. In rats given high dose MeHg+ for 21 days (MeHg+ 5 mg/kg/day with co-administration of 1 mg/kg/day of diphenyl diselenide), MeHg-induced motor deficits and body weight loss worsened [Citation174]. In addition, liver and brain mercury concentrations increased [Citation174]. In a mouse study using Hg+2 and a very high dose of diphenyl diselenide, (HgCl2 4.6 mg/kg/day for 3 days, followed by a single dose of diphenyl diselenide 100 µmol/kg [31.2 mg/kg]), there was 100% lethality [Citation146]. Mice were euthanized at 4 h because the Se dose of 32 mg/kg was previously established to cause 100% death in mice in 5 h [Citation144].
In a rat study using concurrent MeHg+ (at varying doses) and selenium as selenite for 50 weeks (24 µg/kg/day Se, a molar ratio of 18:1), selenium supplementation delayed long-term behavioral and motor effects of MeHg+ exposure compared with MeHg+ poisoned rats receiving no selenium [Citation124]. In this study the dose of MeHg+ and the Hg:Se molar ratio played a significant role in outcome, as measured by onset of aging, run tests, forelimb grip strength, flexion, hind limb crossing, and motor neuron degeneration [Citation124]. In rats receiving MeHg+ 0.4 mg/kg/day and selenium 24 µg/kg/day (6.3:1 Hg:Se molar ratio) there was significant protection from toxicity, however in groups receiving MeHg+ 1.2 mg/kg/day and selenium 24 µg/kg/day (18:1 Hg:Se molar ratio) increasing evidence of neurological and motor injury occurred [Citation124]. In this same study lower dose selenium supplementation with larger Hg:Se molar ratios preformed worse: Hg:Se ratio of 187:1 (MeHg+ 1.2 mg/kg/day with selenium 2.4 µg/kg/day) and Hg:Se ratio of 63:1 (MeHg+ 0.4 mg/kg/day with selenium 2.4 µg/kg/day) [Citation124]. While the dose of MeHg+ was important, the molar ratio was the most predictive of toxicity and outcome with Hg:Se ratios of 187 > 63 > 18.7 > 6.3 [Citation124].
These results following acute co-administration of Hg and Se are not consistent across all studies. In a mouse study of 35 days of MeHg+ (2 mg/kg/day MeHg+ with co-administration of 1 mg/kg/day of diphenyl diselenide), there was evidence of decreased oxidative stress in the brain and liver, with decreased Hg deposition in the cerebrum, cerebellum, kidney and liver when compared with MeHg+ poisoned mice receiving no treatment [Citation119].
Alternate markers of mercury toxicity
Selenium supplementation may initially result in an increase in the blood mercury concentration, which is not an indication for discontinuation of selenium supplementation. We would caution against focusing too heavily on mercury concentrations either in the blood or urine as selenium status appears to be more important in determining patient outcome.
Several alternate markers of mercury toxicity have been suggested to monitor ongoing injury and perhaps response to therapy: 1) neurologic injury – serum neuron-specific enolase (NSE), S100B, and glutamate receptor levels, and 2) evidence of oxidative stress – serum malondiadehyde and urinary malondiadehyde and 8-hydroxy-2-deoxyguanosine [Citation131, Citation133, Citation175–177]. However, further validation of these markers are needed and may not be widely available.
Summary of selenium supplementation
The primary role of selenium supplementation appears to be correction of mercury-induced selenium deficiency state, restoration of critical selenoprotein activity and subsequent reduction of oxidative stress and cellular calcium dyshomeostasis (restoration of selenoprotein control over endoplasmic reticulum release of intracellular calcium). Additive roles of selenium may be redistribution of Hg away from target organs, demethylation of organic mercury and possible sequestration of Hg as Hg:Se complexes. A limitation of selenium supplementation is dose. As the intracellular molar ratio of mercury to selenium increases the efficacy of selenium decreases. In very high doses of mercury poisoning, safe doses of selenium supplementation may not be sufficient to approach a protective equimolar ratio. Attempting to manage with selenium alone using a very high dose of selenium may produce significant adverse events and worsen the patient's condition. A second limitation is that selenium has only a modest effect on increased mercury elimination. However, this increased elimination is from a reduction of total body mercury from target organ tissue redistribution, and not an increase from kidney mercury stores.
Limitations
There are no randomized clinical trials in humans evaluating any therapy for mercury poisoning. The existing widespread recommendations for the use of thiol chelators are based on evaluation of animal studies and human case reports and fail to take into account the mechanism of toxicity of mercury. These recommendations have not been tested in randomized clinical trials. The dosing and duration of therapy for unithiol and succimer are based on different metals (i.e. lead) with different kinetics and target organs. The recommendations for management of mercury poisoning in this review are based on evaluation of advances in understanding of mechanism of mercury toxicity, in vitro studies, animal studies and human case reports, but also lack a randomized clinical trial. Randomized clinical trials are recommended, but are likely to prove difficult due to limited patient populations and ethical reasons.
Conclusions
The primary consequence of mercury poisoning is the creation of a selenium deficient state. Previous management of mercury poisoning focused solely on increased renal elimination of mercury via the thiol chelators, did not address selenium status and consequently has produced limited benefit. A combined approach which focuses on restoration of selenoprotein function, reduction of oxidative stress along with increased mercury elimination is suggested.
Selenium supplementation will help restore selenoprotein concentrations and may allow for reactivation of some of the existing selenoproteins, which will help restore intracellular calcium homeostasis and the cellular redox environment via correction of the mercury-induced selenium deficiency state. Additionally selenium supplementation may increase distribution of mercury away from target organs, increase mercury elimination, and help sequester mercury as the inert Se:Hg colloidal complex. Dosing and duration of therapy for selenium supplementation have not been established. Duration of therapy may be based on patient specific determinants (e.g. clinical severity, chronicity). An initial selenium supplementation of 100 µg/day in asymptomatic patients and 500 µg/day in symptomatic patients for 90 days is recommended and has been used by the authors (see and ). Selenium as sodium selenite and selenomethionine have been used. Continued supplementation should be based on reevaluation of a number of patient factors, including response to treatment, adherence, and the presence and severity of any drug effects that may warrant discontinuance of selenium. The recommendation for 90 days is based on available human reports and appears to be a reasonable time for patient reevaluation [Citation48, Citation131–133]. Further research on dose and duration of therapy of selenium is warranted.
Figure 5. Suggested management guideline for Organic Mercury (Methylmercury, dimethylmercury, etc).
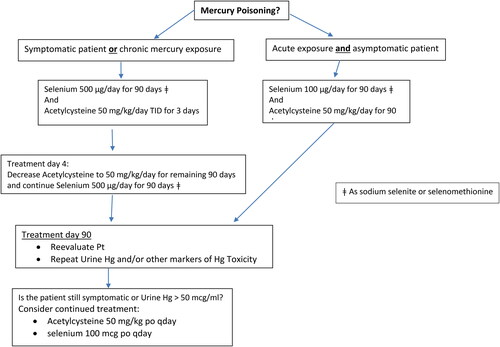
Figure 6. Suggested management guidelines for Inorganic Mercury or Mercury Vapor.
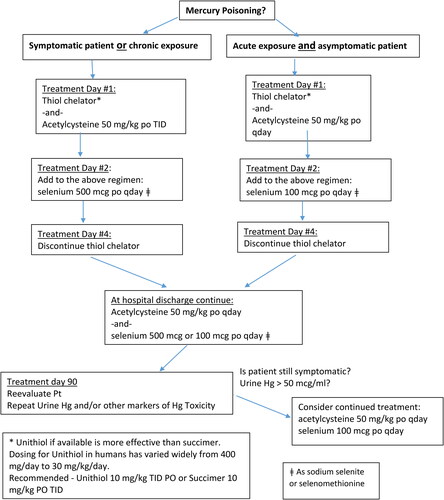
For organic mercury, the ability of acetylcysteine to penetrate the CNS, increase glutathione production, ability to complex with mercury and act as a potent chelator argue that it may be a better choice as a complexing agent. The drawbacks of the thiol chelators are their limited effectiveness with organic mercuries, such as methylmercury and thiomersal, inability to cross the blood-brain barrier, increased excretion of selenium and their short-term effect on increased mercury elimination. Simultaneous use of selenium with unithiol or succimer may initially reduce the efficacy of the thiol chelators and possibly reduce availability of the supplemental selenium. Conversely, simultaneous use of selenium with acetylcysteine appears to be synergistic. For organic mercuries, we recommend selenium supplementation and acetylcysteine for mercury elimination (see ). Dosing and duration of therapy for acetylcyteine supplementation have not been established. Continued supplementation should be based on patient reevaluation.
For inorganic mercury we recommend a combined chelator approach. In the initial few days following inorganic mercury exposure (HgCl2 or mercury vapor), unithiol may be more effective than acetylcysteine in increasing Hg+2 elimination from renal stores and reducing kidney inorganic mercury. For inorganic mercuric salts and elemental mercury, we recommend limiting the use of the thiol chelators and switching to acetylcysteine for continued mercury elimination once the initial phase of renal Hg clearance is achieved. Based on animal and human data this appears to be 2 to 4 days. After switching to acetylcysteine, we recommend continuing acetylcysteine for 90 days. Simultaneous use of selenium with acetylcysteine appears to be synergistic. Unithiol, if available, is more effective than succimer (see ).
Further research is warranted on establishing a more definitive recommendation for selenium dosing and duration of therapy, the form of selenium that may provide best results; investigation of a Hg:Se molar ratio needed to optimize outcomes; and continued examination of the role that thiol-based chelators and acetylcysteine should play in the treatment of mercury poisoning.
Table 1. Evaluation and review of refenced studies.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Spiller HA. Rethinking mercury: the role of selenium in the pathophysiology of mercury toxicity. Clin Toxicol (Phila)). 2018;56(5):313–326.
- Kosnett MJ. The role of chelation in the treatment of arsenic and mercury poisoning. J Med Toxicol. 2013;9(4):347–354.
- Su YJ. Mercury. In: Nelson LS, Howland M, Lewin NA, Smith SW, Goldfrank LR, Hoffman RS, editors. Goldfrank’s toxicologic emergencies. New York (NY): McGraw Hill Medical; 2019. p. 1324–1332.
- Kosnett MJ. Mercury. In: Olsen K, editor. Poisoning & drug overdose. New York (NY): McGraw Hill Education; 2018. p. 305–311.
- Roels HA, Boeckx M, Ceulemans E, et al. Urinary excretion of mercury after occupational exposure to mercury vapour and influence of the chelating agent meso-2,3-dimercaptosuccinic acid (DMSA)). Br J Ind Med. 1991;48(4):247–253.
- Bluhm RE, Bobbit RG, Welch LW, et al. Elemental mercury vapour toxicity, treatment, and prognosis after acute, intensive exposure in chloralkali plant workers. Part I: history, neuropsychological findings and chelator effects. Hum Exp Toxicol. 1992;11(3):201–210.
- Smiechowicz J, Skoczynska A, Nieckula-Szwarc A, et al. Occupational mercury vapour poisoning with a respiratory failure, pneumomediastinum and severe quadriparesis. SAGE Open Med Case Rep. 2017;5:2050313X17695472. http://journals.sagepub.com/doi/10.1177/2050313X17695472
- Planas-Bohne F. The influence of chelating agents on the distribution and biotransformation of methylmercuric chloride in rats. J Pharmacol Exper Therapeutics. 1981;217:500–504.
- Aposhian HV, Morgan DL, Queen SHL, et al. Vitamin C, glutathione, or lipoic acid did not decrease brain or kidney mercury in rats exposed to mercury vapor. J Toxicol Clin Toxicol. 2003;41(4):339–347.
- Aposhian MM, Maiorino RM, Xu Z, et al. Sodium 2,3-dimercapto-1-propanesulfonate (DMSA) treatment does not redistribute lead or mercury to the brain of rats. Toxicology. 1996;109(1):49–55.
- Bridges CC, Joshee L, Zalups RK. Effect of DMPS and DMSA on the placental and fetal disposition of methylmercury. Placenta. 2009;30(9):800–805.
- Nierenberg DW, Nordgren RE, Chang MB, et al. Delayed cerebellar disease and death after accidental exposure to dimethylmercury. N Engl J Med. 1998;338(23):1672–1676.
- Pfab R, Muckter H, Roider G, et al. Clinical course of severe poisoning with thiomersal. J Toxicol Clin Toxicol. 1996;34(4):453–460.
- Kostyniak PJ, Soiefer AI. A methylmercury toxicity model to test for possible adverse effects resulting from chelating agent therapy. J Appl Toxicol. 1984;4(4):206–210.
- Pingree SD, Simmonds PL, Woods JS. Effects of 2,3-dimercapto-1-propanesulfonic acid (DMPS) on tissue and urine mercury levels following prolonged methylmercury exposure in rats. Toxicol Sci. 2001;61(2):224–233.
- Mudan A, Copan L, Wang R, et al. Notes from the field: methylmercury toxicity from a skin lightening cream obtained from Mexico – Califormia, 2019. MMWR Morb Mortal Wkly Rep. 2019;68:1166–1167.
- Dietrich KN, Ware JH, Salganik M, et al. Effect of chelation therapy on the neuropsychological and behavioral development of lead-exposed children after school entry. Pediatrics. 2004;114(1):19–26.
- Baum CR. Treatment of mercury intoxication. Curr Opin Pediatr. 1999;11(3):265–268.
- Aaseth J, Alexander J, Raknerud N. Treatment of mercuric chloride poisoning with dimercaptosuccinic acid and diuretics: preliminary studies. J Toxicol Clin Toxicol. 1982;19(2):173–186.
- Zalups RK, Bridges CC. Relationships between the renal handling of DMPS and DMSA and the renal handling of mercury. Chem Res Toxicol. 2012;25(9):1825–1838.
- Bridges CC, Joshee L, Zalups RK. Multidrug resistance proteins and the renal elimination of inorganic mercury mediated by 2,3-Dimercaptopropane-1-sulfonic acid and meso-2,3-dimercaptosuccinic acid. J Pharmacol Exp Ther. 2008;324(1):383–390.
- Joshi D, Mittal DK, Shukla S, et al. Methylmercury toxicity: amelioration by selenium and water-soluble chelators as N-acetyl cysteine and dithiothreitol. Cell Biochem Funct. 2014;32(4):351–360.,
- Joshi D, Mittal DK, Shukla S, et al. N-acetyl cysteine and selenium protects mercuric chloride-induced oxidative stress and antioxidant defense system in liver and kidney of rats: a histopathological approach. J Trace Elem Med Biol. 2014;28(2):218–226.,
- Joshi D, Mittal DK, Bhadauria M, et al. Role of micronutrients against dimethylmercury intoxication in male rats. Environ Toxicol Pharmacol. 2010;29(2):97–103.
- Rooney JPK. The role of thiols, dithiols, nutritional factors and interacting ligands in the toxicology of mercury. Toxicology. 2007;234(3):145–156.
- Ballatori N, Wang W, Lieberman MW. Accelerated methylmercury elimination in γ-glutamyl transpeptidase-deficient mice. Am J Pathol. 1998;152:1049–1055.
- Ballatori N, Lieberman MW, Wang W. N-acetylcysteine as an antidote in methylmercury poisoning. Environ Health Perspect. 1998;106(5):267–271.
- Girardi G, Elias MM. Effect of different renal glutathione levels on renal mercury disposition and excretion in the rat. Toxicology. 1993;81(1):57–67.
- Zalups RK, Ahmad S. Transport of N-acetylcysteine s-conjugates of methylmercury in Madin-Darby canine kidney cells stably transfected with human isoform of organic anion transporter 1. J Pharmacol Exp Ther. 2005;314(3):1158–1168.
- Koh AS, Simmons-Willis TA, Pritchard JB, et al. Identification of a mechanism by which the methylmercury antidotes N-acetylcysteine and dimercaptopropanesulfonate enhance urinary metal excretion: transport by the renal organic Anion transporter-1. Mol Pharmacol. 2002;62(4):921–926.
- Kaur P, Aschner M, Syversen T. Glutathione modulation influences methyl mercury induced neurotoxicity in primary cell cultures of neurons and astrocytes. Neurotoxicology. 2006;27(4):492–500.
- Falluel-Morel A, Lin L, Sokolowski K, et al. N-Acetyl cysteine treatment reduces mercury-induced neurotoxicity in the developing rat hippocampus. J Neurosci Res. 2012;90(4):743–750.
- Madejczyk MS, Aremu DA, Simmons-Willis TA, et al. Accelerated urinary excretion of methylmercury following administration of its antidote n-acetylcysteine requires MRP2/ABCC2, the apical multidrug resistance-associated protein. J Pharmacol Exp Ther. 2007;322(1):378–384.
- Aremu DA, Madejczyk MS, Ballatori N. N-acetylcysteine as a potential antidote and biomonitoring agent of methylmercury exposure. Environ Health Perspect. 2008;116(1):26–31.
- Ottenwalder H, Simon P. Differential effect of N-acetylcysteine on excretion of the metals Hg, Cd, Pb and Au. Arch Toxicol. 1987;60(5):401–402.
- Usuki F, Ishiura S. Expanded CTG repeats in myotonin protein kinase increase susceptibility to oxidative stress. Clin Neurosci. 1998;9:2291–2296.
- Becker A, Soliman FFA. The role of intracellular glutathione in inorganic mercury-induced toxicity in neuroblastoma cells. Neurochem Res. 2009;34(9):1677–1684.
- Girardi G, Elias MM. Effectiveness of N-acetylcysteine in protecting against mercuric chloride-induced nephrotoxicity. Toxicology. 1991;67(2):155–164.
- Simpson PG, Hopkins TE, Haque R. Binding of methylmercury chloride to the model peptide, N-Acetyl-L-Cysteine. A proton magnetic resonance study. J Phys Chem. 1973;77(19):2282–2285.
- Dutczak WJ, Ballatori N. Transport of glutathione-methylmercury across liver canalicular membranes on reduced glutathione carriers. J Biol Chem. 1994;269(13):9746–9751.
- Fujiyama J, Hirayama K, Yasutake A. Mechanism of methylmercury efflux from cultured astrocytes. Biochem Pharmacol. 1994;47(9):1525–1530.
- Rush T, Liu X, Nowakowski AB, et al. Glutathione-mediated neuroprotection against methylmercury neurotoxicity in cortical culture is dependent on MRP1. Neurotoxicology. 2012;33(3):476–481.
- Livardjani F, Ledig M, Kopp P, et al. Lung and blood superoxide dismutase activity in mercury vapor exposed rats: effect of N-acetylcysteine treatment. Toxicology. 1991;66(3):289–295.
- Yuntao F, Chenjia G, Panpan Z, et al. Role of autophagy in methylmercury-induced neurotoxicity in rat primary astrocytes. Arch Toxicol. 2016;90(2):333–345.
- Napp LC, Moelgen C, Wegner F, et al. Multimodal elimination for intoxication with a lethal dose of organic mercury. Case Rep Crit Care. 2019; 2019:4275918.
- Ho BSJ, Lin J-l, Huang C-C, et al. Mercury vapor inhalation from Chinese red (Cinnabar). J Toxicol Clin Toxicol. 2003;41(1):75–78.
- Benz MR, Lee SH, Kellner L, et al. Hyperintense lesions in brain MRI after exposure to a mercuric chloride-containing skin whitening cream. Eur J Pediatr. 2011;170(6):747–750.
- Spiller HA, Hays HL, Burns G, et al. Severe elemental mercury poisoning managed with selenium and N-acetylcysteine administration. Toxicol Comm. 2017;1(1):24–28.
- Oz SG, Tozlu M, Yalcin SS, et al. Mercury vapor inhalation and poisoning of a family. Inhal Toxicol. 2012;24(10):652–658.
- Zhang J. Clinical observations in ethyl mercury chloride poisoning. Am J Ind Med. 1984;5(3):251–258.
- Bradberry SM, Sheehan TMT, Barraclough CR, et al. DMPS can reverse the features of severe mercury vapor-induced neurological damage. Clin Toxicol (Phila)). 2009;47(9):894–898.
- Gattineni J, Weiser S, Becker AM, et al. Mercury intoxication: lack of correlation between symptoms and levels. Clin Pediatr (Phila)). 2007;46(9):844–846.
- Torres AD, Rai AN, Hardiek ML. Mercury intoxication and arterial hypertension: report of two patients and review of the literature. Pediatrics. 2000;105(3):E34.
- Laurans M, Brouard J, Arion A, et al. Familial mercury intoxication presenting with cardiovascular abnormalities and acrodynia. Acta Paediatr. 2001;90(5):593–594.
- Mercer JJ, Bercovitch L, Muglia JJ. Acrodynia and hypertension in a young girl secondary to elemental mercury toxicity acquired in the home. Pediatr Dermatol. 2012;29(2):199–201.
- Yeats KO, Mortenson ME. Acute and chronic neuropsychological consequences of mercury vapor poisoning in two early adolescents. J Clin Exp Neuropysch. 1994;16:209–222.
- Vallant B, Deutsch J, Muntean M, et al. Intravenous injection of metallic mercury: case report and course of mercury during chelation therapy with DMPS. Clin Toxicol. 2008;46(6):566–569.
- Toet AE, van Dijk A, Savelkoul TJF, et al. Mercury kinetics in a case of severe mercuric chloride poisoning treated with dimercapto-1-propane sulphonate (DMPS). Hum Exp Toxicol. 1994;13(1):11–16.
- Clarkson TW, Magos L, Cox C, et al. Tests of efficacy of antidotes for removal of methylmercury in human poisoning during the Iraq outbreak. J Pharmacol Exp Therap. 1981;218:74–83.
- Beck C, Krafchik B, Traubici J, et al. Mercury intoxication: it still exists. Pediatr Dermatol. 2004;21(3):254–259.
- Brannan EH, Su S, Alverson BK. Elemental mercury poisoning presenting as hypertension in a young child. Pediatr Emerg Care. 2012;28:812–814.
- Fayez I, Paiva M, Thompson M, et al. Toxicokinetics of mercury elimination by succimer in twin toddlers. Pediatr Drugs. 2005;7:397–400.
- Pierce PE, Thompson JF, Likowsky WH, et al. Alkyl mercury poisoning in humans. J Am Med Ass. 1972;220(11):1439–1442.
- Weinstein M, Bernstein S. Pink ladies: mercury poisoning in twin girls. Can Med Assoc J. 2003;168:201.
- Carter M, Abdi A, Naz F, et al. A mercury toxicity case complicated by hyponatremia and abnormal endocrinological test results. Pediatrics. 2017;140:e20161402.
- Magos L. Three cases of methylmercury intoxication which eluded correct diagnosis. Arch Toxicol. 1998;72(11):701–705.
- Bluhm RE, Breyer JA, Bobbit RG, et al. Elemental mercury vapour toxicity, treatment, and prognosis after acute, intensive exposure in chloralkali plant workers. Part II: hyperchloraemia and genitourinary symptoms. Hum Exp Toxicol. 1992;11(3):211–215.
- Lund ME, Banner W, Clarkson TW, et al. Treatment of acute methylmercury ingestion by hemodialysis with n-acetylcysteine (Mucomyst) infusion and 2,3-dimercaptopropane sulfonate. J Toxicol Clin Toxicol. 1984;22(1):31–49.
- Koch M, Trapp R. Ethyl mercury poisoning during a protein A immunoadsorption treatment. Am J Kidney Dis. 2006;47(2):e31–e34.
- Forman J, Moline J, Cernichiari E, et al. A cluster of pediatric metallic mercury exposure cases treated with meso-2,3-dimercaptosuccinic acid (DMSA). Environ Health Perpect. 2000;108(6):575–577.
- Dargan PL, Giles LJ, Wallace CI, et al. Case report: severe mercuric sulphate poisoning treated with 2,3-dimercaptopropane-1-sulphonate and haemodiafiltration. Crit Care. 2003;7(3):R1–R6.
- Ly BT, Williams SR, Clark RF. Mercuric oxide poisoning treated with whole-bowel irrigation and chelation therapy. Ann Emerg Med. 2002;39(3):312–315.
- Houeto P, Sandouk P, Baud FJ, et al. Elemental mercury vapour toxicity: treatment and levels in plasma and urine. Hum Exp Toxicol. 1994;13(12):848–852.
- Pelclova D, Vlckova S, Bezdicek O, et al. Is chelation therapy efficient for the treatment of intravenous metallic mercury intoxication? Basic Clin Pharmacol Toxicol. 2017;120(6):628–633.
- Gonzalez-Ramirez D, Zuniga-Charles M, Narro-Juarez A, et al. DMPS (2,3-dimercaptopropane-1-sulfonate, dimaval) decreases the body burden of mercury in humans exposed to mercurous chloride. J Pharmacol Exp Therap. 1998;287(2):8–12.
- Bjelosevic M, Fabianova M, Olejnik P, et al. Pediatric mercury intoxication mimicking pheochromcytoma. Balkan Med J. 2018;35:451–452.
- Buchet JP, Lauwerys RR. Influence of 2,3-dimercaptopropane-1-sulfonate and dimercaptosuccinic acid on the mobilization of mercury from tissues of rats pretreated with mercuric chloride, phenylmercury acetate or mercury vapors. Toxicology. 1989;54(3):323–333.
- Nerudová J, Cábelková Z, Frantík E, Lukás E, et al. Mobilization of mercury by DMPS in occupationally exposed workers and in model experiments on rats: evaluation of body burden. Int J Occup Med Environ Health. 2000;13(2):131–146.
- Lundgren KD, Swensson A, Ulfvarson U. Studies in humans on the distribution of mercury in the blood and the excretion in urine after exposure to different mercury compounds. Scand J Clin Lab Invest. 1967;20(2):164–166.
- Clarkson TW, Magos L. The toxicology of mercury and its chemical compounds. Crit Rev Toxicol. 2006;36(8):609–662.
- Vahter ME, Mottet NK, Friberg LT, et al. Demethylation of methylmercury in different brain sites in macaca fascicularis monkeys during long term subclinical methylmercury exposure. Toxicol App Pharmacol. 1995;134(2):273–284.
- Burbacher TM, Shen DD, Liberato N, et al. Comparison of blood and brain mercury levels in infant monkeys exposed to methylmercury or vaccines containing thimerosal. Environ Health Perspect. 2005;113(8):1015–1021.
- Joshi D, Mittal DK, Shrivastava S, et al. Protective role of thiol chelators against dimetylmercury induced toxicity in male rats. Bull Environ Contam Toxicol. 2010;84(5):613–617.
- Dorea JG, Farina M, Rocha JBT. Toxicity of ethylmercury (and Thimerosal): a comparison with methylmercury. J Appl Toxicol. 2013;33(8):700–711.
- Lowel JA, Burgess S, Shenoy S, et al. Mercury poisoning associated with hepatitis-B immunoglobulin. Lancet. 1996;347(8999):480.
- Carneiro MFH, Souza JMO, Grotto D, et al. A systematic study of the disposition and metabolism of mercury species in mice after exposure to low levels of thimerosal (ethylmercury). Environ Res. 2014;134:218–227.
- Kern JK, Geier DA, Homme KG, Geier MR. Examining the evidence that ethylmercury crosses the blood-brain barrier. Environ Toxicol Pharmacol. 2020;74:103312.
- Zieminska E, Toczylowska B, Stafiej A, et al. Low molecular weight thiols reduce thimerosal neurotoxicity in vitro: modulation by proteins. Toxicology. 2010;276(3):154–163.
- Solis MT, Yuen E, Cortez PS, et al. Family poisoned by mercury vapor inhalation. Am J Emerg Med. 2000;18(5):599–602.
- Eyer F, Felgenhauer N, Pfab R, et al. Neither DMPS nor DMSA is effective in quantitative elimination of elemental mercury after intentional IV injection. Clin Toxicol (Phila)). 2006;44(4):395–397.
- Magos L. The effects of dimercaptosuccinic acid on the excretion and distribution of mercury in rats and mice treated with mercuric chloride and methylmercury chloride. Br J Pharmacol. 1976;56(4):479–484.
- Gabard B. Treatment of methylmercury poisoning in the rat with sodium 2,3-dimercaptopropane-l-sulfonate: influence of dose and mode of administration. Toxicol Appl Pharmacol. 1976;38(2):415–424.
- Ewan KBR, Pamphlett R. Increased inorganic mercury in spinal motor neurons following chelating agents. Neurotoxicol. 1996;17:343–350.
- Garza-Ocanas L, Torres-Alanis O, Pineryro-Lopez A. Urinary mercury in twelve cases of cutaneous mecurous chloride (Calomel) exposure: effect of sodium 2,3-dimercaptopropane-1-sulfonate (DMPS) therapy. Clin Toxicol. 1997;35:653–655.
- Torres-Alanís O, Garza-Ocañas L, Pineyro-Lopez A. Evaluation of urinary excretion after administration of 2,3-dimercapto-1-propane sulfonic acid to occupationally exposed men. Clin Toxicol. 1995;33(6):717–720.
- Hurlbut KM, Maiorino RM, Mayersohn M, et al. Determination and metabolism of dithiol chelating agents. XVI: pharmacokinetics of 2,3-dimercapto-1-propanesulfonate after intravenous administration to human volunteers. J Pharmacol Exp Ther. 1994; 268(2):662–668.
- Ganther HE. Modification of methylmercury toxicity and metabolism by selenium and vitamin E: possible mechanisms. Environ Health Perspect. 1978;25:71–76.
- Parizek J, Ostadalova I. The protective effect of small amounts of selenite in sublimate intoxication. Experimentia. 1967;23:142–143.
- Li Y, Fan Y, Zhao J, et al. Elevated mercury bound to serum proteins in methyl mercury poisoned rats after selenium treatment. Biometals. 2016;29(5):893–903.
- Ralston NCV, Ralston CR, Blackwell JL, et al. Dietary and tissue selenium in relation to methylmercury toxicity. Neurotoxicol. 2008;29(5):802–811.
- Seppanen K, Laatikainen R, Salonen JT, et al. Mercury-binding capacity of organic and inorganic selenium in rat blood and liver. Biol Trace Elem Res. 1998;65(3):197–210.
- Li X, Yin D, Chen Q, et al. Dietary selenium protect against redox-mediated immune suppression induced by methylmercury exposure . Food Chem Toxicol. 2014;72:169–177.
- Stillings BR, Lagally H, Bauersfeld P, et al. Effects of cysteine, selenium and fish protein on toxicity and metabolism of methylmercury in rats. Toxicol Pharmacol. 1974;30(2):243–254.
- Ralston NVC, Raymond LJ. Dietary selenium's protective effects against methylmercury toxicity. Toxicology. 2010;278(1):112–123.
- Branco V, Carvalho C. The thioredoxin system as a target for mercury compounds. Biochemica Biophysic Acta Gen Subj. 2019;1863(12):129255.
- Carvalho CML, Chew EH, Hashemy SI, et al. Inhibition of the human thioredoxin system. A molecular mechanism of mercury toxicity. J Biol Chem. 2008;283(18):11913–11923.
- Branco V, Godinho-Santos A, Gonçalves J, et al. Mitochondrial thioredoxin system as a primary target for mercury compounds. Toxicol Lett. 2014;229(Suppl):S57–S58.
- Branco V, Lu J, Holmgren A, et al. Effect of mercury compounds over the redox state of thioredoxin 1 and 2 and its relation with glutathione levels and glutaredoxin activity in human neuroblastoma cells. Toxicol Lett. 2015;238(2):S314.
- Sugiura Y, Tamai Y, Tanaka H. Selenium protection against mercury toxicity: high binding affinity of methylmercury by selenium-containing ligands in comparison with sulfur-containing ligands. Bioorganic Chem. 1978;9(2):167–180.
- Grumolato L, Ghzili H, Montero-Hadjadje M, et al. Selenoprotein T is a PACAP-regulated gene involved in intracellular Ca2+ mobilization and neuroendocrine secretion. FASEB J. 2008;22(6):1756–1768.
- Raymond LJ, Ralston NVC. Mercury: selenium interactions and health implications. Seychelles Med Den J. 2004;7:72–77.
- Wang C, Li R, Huang Y, et al. Selenoprotein K modulate intracellular free Ca2+ by regulating expression of calcium homoeostasis endoplasmic reticulum protein. Biochem Biophys Res Commun. 2017;484(4):734–739.
- Branco V, Godinho-Santos A, Goncalves J, et al. Mitochondrial thioredoxin reductase inhibition, selenium status, and NRF-2 activation are determinant factors modulating the toxicity of mercury compounds. Free Radic Biol Med. 2014;73:95–105.
- Glaser V, Moritz B, Schmitz A, et al. Protective effects of diphenyl diselenide in a mouse model of brain toxicity. Chem Biol Interact. 2013;206(1):18–26.
- Cabanero AL, Madrid Y, Camara C. Selenium long-term administration and its effect on mercury toxicity. J Agric Food Chem. 2006;54(12):4461–4468.
- Khan MAK, Wang F. Mercury-selenium compounds and their toxicological significance: toward a molecular understanding of the mercury-selenium antagonism. Environ Toxicol Chem. 2009;28(8):1567–1577.
- de Freitas AS, Funck VR, Rotta MDS, et al. Diphenyl diselenide, a simple organoselenium compound, decreases methylmercury-induced cerebral, hepatic and renal oxidative stress and mercury deposition in adult mice. Brain Res Bull. 2009;79(1):77–84.
- Dalla Corte CL, Soares FAA, Aschner M, et al. Diphenyl Diselenide prevents methylmercury-induced mitochondrial dysfunction in rat liver slices. Tetrahedron. 2012;68(51):10437–10444.,
- Glaser V, Nazari EM, Muller YMR, et al. Effects of inorganic selenium administration in methylmercury-induced neurotoxicity in mouse cerebral cortex. Int J Dev Neurosci. 2010;28(7):631–637.
- Gajdosechova Z, Lawan MM, Urgast DS, et al. In vivo formation of natural HgSe nanoparticles in the liver and brain of pilot whales. Sci Rep. 2016;6:34361.
- Korbas M, O’Donoghue JL, Watson GE, et al. The chemical nature of mercury in human brain following poisoning or environmental exposure. ACS Chem Neurosci. 2010;1(12):810–818.
- Bulato C, Bosello V, Ursini F, et al. Effect of mercury on selenium utilization and selenoperoxidase activity in LNCaP cells. Free Radic Biol Med. 2007;42(1):118–123.
- Li Y, Ge Y, Wang R, et al. Nanoelemental selenium alleviated the mercury load and promoted the formation of high-molecular-weight mercury- and selenium-containing proteins in serum samples from methylmercury-poisoned rats. Ecotoxicol Environ Saf. 2019;169:128–133.
- Heath JC, Banna KM, Reed MN, et al. Dietary selenium protects against selected signs of aging and methylmercury exposure. Neurotoxicology. 2010;31(2):169–179.
- Carvalho CML, Lu JL, Arner ES, et al. Effects of selenite and chelating agents on mammalian thioredoxin reductase inhibited by mercury: implications for treatment of mercury poisoning. FASEB J. 2011;25(1):370–381.
- Nygaard SP, Hansen JC. Mercury-selenium interaction at concentrations of selenium and of mercury vapours as prevalent in nature. Bull Environ Contam Toxicol. 1978;20(1):20–23.
- Grotto D, Barcelos GRM, Valentini J, et al. Low level of methylmercury induce DNA damage to rats: protective effects of selenium. Arch Toxicol. 2009;83(3):249–254.
- Anderson O, Nielson JB. Effects of simultaneous low-level dietary supplementation with inorganic and organic selenium on whole-body, blood and organ levels of toxic metals in mice. Environ Health Perpect. 1994;102:321–324.
- Glaser V, Martins R. d P, Vieira AJH, et al. Diphenyl diselenide administration enhances cortical mitochondrial number and activity by increasing hemeoxygenase type 1 content in a methylmercury-induced neurotoxicity mouse model. Mol Cell Biochem. 2014;390(1-2):1–9.
- Newland MG, Reed MN, LeBlanc A, et al. Brain and blood mercury and selenium after chronic and developmental exposure to methylmercury. Neurotoxicology. 2006;27(5):710–720.
- Li YF, Chen C, Li B, et al. Simultaneous speciation of selenium and mercury in human urine samples from long term mercury exposed populations with supplementation of selenium-enriched yeast by HPLC ICP-MS. J Anal Atomic Spectrom. 2007;22(8):925.
- Li YF, Dong Z, Chen C, et al. Organic selenium supplementation increases mercury excretion and decreases oxidative damage in long-term mercury-exposed residents from Wanshan, China. Environ Sci Technol. 2012;46(20):11313–11318.
- Draz E, El-Kelany R, El-Namir T, et al. Role of selenium and vitamin E in occupational exposure to heavy metals (mercury, lead and cadmium): impact of working in lamp factory. Mansoura J Forensic Med Clin Toxicol. 2009;17:87–107.
- Jing H, Yu-Feng LI, Shen Y, et al. Effects of sodium selenite on methylmercury poisoned rats. J Mountain Agric Bio. 2104;32:55–60.
- Liu Y, Zhang W, Zhao J, et al. Selenoprotein P as the major transporter for mercury in serum from methylmercury-poisoned rats. J Trace Elem Med Biol. 2018;50:589–595.
- El Asar HM, Mohammed EA, Aboulhoda BE, et al. Selenium protection against mercury neurotoxicity: modulation of apoptosis and autophagy in the anterior pituitary. Life Sci. 2019;231:116578.
- Wang H, Chen B, He M, et al. Selenocystine against methyl mercury cytotoxicity in HepG2 cells. Sci Rep. 2017;7(1):147.
- Liu Y, Ji J, Zhang W, et al. Selenium modulated gut flora and promoted decomposition of methylmercury in methylmercury poisoned rats. Ecotoxicol Environ Saf. 2019;185:109720.
- Lemire M, Fillion M, Frenette B, et al. Selenium from dietary sources and motor functions in the Brazilian Amazon. Neurotoxicology. 2011;32(6):944–953.
- Nakamura M, Hachiya N, Murata KY, et al. Methylmercury exposure and neurological outcomes in Taiji residents accustomed to consuming whale meat. Environ Inter. 2014;68:25–32.
- Burk RF, Norsworthy BK, Hill KE, et al. Effects of chemical form of selenium on plasma biomarkers in a high-dose human supplementation trial. Cancer Epidemiol Biomarkers Prev. 2006;15(4):804–810.
- MacFarquhar JK, Broussard DL, Melstrom P, et al. Acute selenium toxicity associated with a dietary supplement. Arch Intern Med. 2010;170(3):256–261.
- Li P, Li Y, Feng X. Mercury and selenium interactions in human blood in Wanshan mercury mining area, China. Sci Total Environ. 2016;573:376–381.
- Brandao R, Moresco RN, Belle LP, et al. Diphenyl diselenide potentiates nephrotoxicity induced by mercuric chloride in mice. J Appl Toxicol. 2011;31(8):773–782.
- Hill CH. Reversal of selenium toxicity in chicks by mercury, copper, and cadmium. J Nutr. 1974;104(5):593–598.
- Rush T, Hjelmhaug J, Lobner D. Effects of chelators on mercury, iron, and lead neurotoxicity in cortical culture. Neurotoxicology. 2009;30(1):47–51.
- Agency for Toxic Substances and Disease Registry (ATSDR). Toxicologic profile for selenium. Atlanta (GA): US Department of Health and Human Services, Public Health Service; 2003.
- Alkazemi D, Egeland GM, Roberts LJ, et al. New insights regarding tissue Se and Hg interactions on oxidative stress from plasma IsoP and IsoF measures in the Canadian Inuit population. J Lipid Res. 2013;54(7):1972–1979.
- Lemire M, Philibert A, Fillion M, et al. No evidence of selenois from a selenium rich diet in the Brazilian amazon. Eviron Intern. 2012;40:128–136.
- Moller-Madsen B, Danscher G. Localization of mercury in CNS of the rat. IV. The effect of selenium on orally administered organic and inorganic mercury. Toxicol Appl Pharmacol. 1991;108(3):457–473.
- Simmons-Willis TA, Koh AS, Clarkson TW, et al. Transport of a neurotoxicant by molecular mimicry: the methylmercury-L-cysteine complex is a substrate for human L-type large neutral amino acid transporter (LAT) 1 and LAT2. Biochem J. 2002;367(Pt 1):239–246.
- Aschner M, Eberle NB, Goderie S, et al. Methylmercury uptake in rat primary astrocyte cultures: the role of the neutral amino acid transport system. Brain Res. 1990;521(1-2):221–228.
- Khan MAK, Wang F. Chemical demethylation of methylmercury by selenoamino acids. Chem Res Toxicol. 2010;23(7):1202–1206.
- Lind B, Friberg L, Nylander M. Preliminary studies on methylmercury biotransformation and clearance in the brain of primates: II. Demethylation of mercury in brain. J Trace Elem Exp Med. 1988;1:49–56.
- Sakamoto M, Itai T, Nakamira M, et al. Detoxification of methylmercury by formation of mercury selenide in muscle of toothed-whale. Toxicol Lett. 2015;238(2):S95.
- Sakamoto M, Yasutake A, Kakita A, et al. Selenomethionine protects against neuronal degeneration by methylmercury in developing rat cerebrum. Environ Sci Technol. 2013;47:2861–2868.
- Kosta L, Byrne AR, Zelenko V. Correlation between selenium and mercury in man following exposure to inorganic mercury. Nature. 1975;254(5497):238–239.
- Dalla Corte CL, Ramos A, Dos Santos CMM, et al. Selenium and mercury levels in rat liver slices co-treated with diphenyl diselenide and methylmercury. Biometals. 2016;29(3):543–550.
- Kuras R, Reszka E, Wieczorek E, et al. Biomarkers of selenium status and antioxidant effect in workers occupationally exposed to mercury. J Trace Elem Med Biol. 2018;49:43–50.
- Chen C, Yu H, Zhao J, et al. The roles of serum selenium and selenoproteins on mercury toxicity in environmental and occupational exposure. Environ Health Perspect. 2006;114(2):297–301.
- Penglase S, Hamre K, Ellingsen S. Selenium prevents downregulation of antioxidant selenoprotein genes by methylmercury. Free Radic Biol Med. 2014;75:95–104.
- Carvalho MC, Franco JL, Ghizoni H, et al. Effects of 2,3-dimercapto-1-propanesulfonic acid (DMPS) on methylmercury-induced locomotor deficits and cerebellar toxicity in mice. Toxicology. 2007;239(3):195–203.
- Kim YJ, Chai YG, Ryu JC. Selenoprotein W as molecular target of methylmercury in human neuronal cells is down-regulated by GSH depletion. Biochem Biophys Res Commun. 2005;330(4):1095–1102.
- Iwata H, Masukawa T, Kito H, et al. Degradation of methylmercury by selenium. Life Sci. 1982;31(9):859–866.
- Yoneda S, Suzuki KT. Detoxification of mercury by selenium by binding of equimolar Hg-Se complex to a specific plasma protein. Toxicol Appl Pharmacol. 1997;143(2):274–280.
- Glynn AW, Lind Y. Effect of long-term sodium selenite supplementation on levels and distribution of mercury in blood, brain and kidneys of methyl mercury-exposed female mice. Pharmacol Toxicol. 1995;77(1):41–47.
- Garcia-Sevillano MA, Rodriquez-Moro G, Garcia-Barrera T, et al. Biological interactions between mercury and selenium in distribution and detoxification process in mice under controlled exposure. Effects on selenoprotein. Chemico Biol Interact. 2015;229:82–90.
- Juresa D, Blanusa M, Kostial K. Simultaneous administration of sodium selenite and mercuric chloride decreases efficacy of DMSA and DMPS in mercury elimination in rats. Toxicol Lett. 2005;155(1):97–102.
- Brandao R, Borges LP, Nogueira CW. Concomitant administration of sodium 2,3-dimercapto-1-propanesulphonate (DMPS) and diphenyl diselenide reduces effectiveness of DMPS in restoring damage induced by mercuric chloride in mice. Food Chem Toxicol. 2009;47(8):1771–1778.
- Torres-Alanis O, Garza-Ocanas L, Bernal MA, et al. Urinary excretion of trace elements in humans after sodium 2,3-dimercaptopropane-1-sulfonate challenge test. J Toxicol Clin Toxicol. 2000;38(7):697–700.
- Paul M, Mason R, Edwards R. Effect of potential antidotes on the acute toxicity, tissue disposition and elimination of selenium in rats. Res Commun Clin Pathol Pharmacol. 1989;66:441–450.
- Civil IDS, McDonald MJA. Acute selenium poisoning: case report. N Z Med J. 1978;87(612):354–356.
- Das K, Dupont A, de Pauw-Gillet C, et al. Absence of selenium protection against methylmercury toxicity in harbour seal leucocytes in vitro. Mar Pollut Bull. 2016;108(1-2):70–76.
- Dalla Corte CL, Wagner C, Sudati JH, et al. Effects of diphenyl diselenide on methylmercury toxicity in rats. Biomed Res Int. 2013; 2013:983821.
- Yilmaz FM, Yilmaz H, Tutkun S, et al. Serum biochemical markers of central nervous system damage in children with acute elemental mercury intoxication. Clin Toxicol. 2014;52(1):32–38.
- Yardan T, Erenler AK, Baydin A, et al. Usefulness of S100B protein in neurological disorders. J Pak Med Assoc. 2011;61(3):276–281.
- Costa A, Branca V, Pigatto PD, et al. Pediatric mercury poisoning, brain MRI, and white matter hyperintensities. Eur J Pediatr. 2011;170(5):677–677.