
ABSTRACT
Background
Chronic postsurgical pain (CPSP) in children remains an important problem with no effective preventive or therapeutic strategies. Recently, genomic underpinnings explaining additional interindividual risk beyond psychological factors have been proposed.
Aims
We present a comprehensive review of current preclinical and clinical evidence for genetic and epigenetic mechanisms relevant to pediatric CPSP.
Methods
Narrative review.
Results
Animal models are relevant to translational research for unraveling genomic mechanisms. For example, Cacng2, p2rx7, and bdnf mutant mice show altered mechanical hypersensitivity to injury, and variants of the same genes have been associated with CPSP susceptibility in humans; similarly, differential DNA methylation (H1SP) and miRNAs (miR-96/7a) have shown translational implications. Animal studies also suggest that crosstalk between neurons and immune cells may be involved in nociceptive priming observed in neonates. In children, differential DNA methylation in regulatory genomic regions enriching GABAergic, dopaminergic, and immune pathways, as well as polygenic risk scores for enhanced prediction of CPSP, have been described. Genome-wide studies in pediatric CPSP are scarce, but pathways identified by adult gene association studies point to potential common mechanisms.
Conclusions
Bench-to-bedside genomics research in pediatric CPSP is currently limited. Reverse translational approaches, use of other -omics, and inclusion of pediatric/CPSP endophenotypes in large-scale biobanks may be potential solutions. Time of developmental vulnerability and longitudinal genomic changes after surgery warrant further investigation. Emergence of promising precision pain management strategies based on gene editing and epigenetic programing emphasize need for further research in pediatric CPSP-related genomics.
RÉSUMÉ
Contexte: La douleur chronique post-chirurgicale (DCPC) chez l'enfant reste un problème important pour lequel il n’y a pas de stratégies préventives ou thérapeutiques efficaces. Récemment, des fondements génomiques expliquant des risques interindividuels additionnels, au-delà des facteurs psychologiques, ont été proposés.
Objectifs: Nous présentons une revue compléte des données probantes précliniques et cliniques actuelles pour les mécanismes génétiques et épigénétiques pertinents en matiére de DCPC pédiatrique.
Méthodes: Revue narrative.
Résultats: Les modéles animaux sont pertinents pour la recherche translationnelle afin de déchiffrer les mécanismes génomiques. Par exemple, les souris mutantes Cacng2, p2rx7 et bdnf présentent une hypersensibilité mécanique altérée à des lésions et des variantes des mêmes génes ont été associées à la sensibilité à la DCPC chez l’humain; de même, la méthylation différentielle de l'ADN (H1SP) et les miARN (miR-96/7a) ont montré des implications translationnelles. Des études menées sur des animaux indiquent également que la diaphonie entre les neurones et les cellules immunitaires peut être impliquée dans l'amorçage nociceptif observé chez les nouveau-nés. Chez les enfants, la méthylation différentielle de l'ADN dans les régions génomiques régulatrices enrichissant les voies GABAergiques, dopaminergiques et immunitaires, ainsi que des scores de risque polygénique pour une prédiction améliorée de la PCSP, ont été décrits. Les études pangénomiques en matiére de DCPC pédiatrique sont rares, mais les voies identifiées par les études d'association de génes chez l'adulte indiquent de possibles mécanismes communs.
Conclusions: La recherche en génomique du laboratoire au patient dans le cadre de la DCPC pédiatrique est actuellement limitée. Les approches translationnelles inversées, l’utilisation d'autres -omiques et l’inclusion d'endophénotypes pédiatriques/DCPC dans les biobanques à grande échelle peuvent être des solutions possibles. La durée de la vulnérabilité développementale et des changements génomiques longitudinaux aprés la chirurgie justifie des recherches plus approfondies. L'émergence de stratégies de précision prometteuses basées sur lé'dition de génes et la programmation épigénétique pour la prise en charge de la douleur font valoir la nécessité de poursuivre les recherches sur la génomique pédiatrique liée à la DCPC.
Introduction
Chronic postsurgical pain (CPSP) has recently been recognized as an entity in the International Classification of Diseases, 11th Revision.1 It is being increasingly studied in pediatric cohorts where the incidence is reported as 14.5% to 38%.Citation1,Citation2 Importantly, up to 33% of preterm babies require surgery, and a higher proportion undergo painful procedures in the neonatal intensive care unit (NICU). Major surgery within the first 3 months of life has been associated with increased pain sensitivity and analgesic requirements with subsequent surgeries compared with infants with no prior surgery, and time spent in the NICU has been linked with increased nociceptive sensitivity in school-aged children, possibly due to repeated painful stimuli received as neonates.Citation3,Citation4 With a high likelihood of hypersensitivity later in life,Citation5 the reported incidence of CPSP in children is likely just the tip of the iceberg for this phenomenon and is only likely to increase in the future.Citation6 The presence of preoperative pain and acute postoperative pain intensity (poorly controlled pain in the immediate and subacute periods) have been identified as risk factors for the development of CPSP,Citation7,Citation8 so much of the early research in this field focused on understanding the mechanisms underlying acute pain after surgery as a way of preventing the transition to CPSP. Psychosocial factors such as anxiety sensitivity,Citation7,Citation9 perioperative factors such as surgical duration,Citation7 and parent–child interactions,Citation3 Citation2,Citation10 have been shown to have both positive and negative influences on CPSP development in children.Citation11 These factors have ~72% accuracy in explaining 16% of interindividual CPSP susceptibility variability in children undergoing spine fusion.Citation7 The heritability of chronic pain susceptibility is estimated at ~50%Citation12–14 based on family and twin studies, with genetic effects accounting for 12% to 60% response variability to experimental painCitation15 and chronic pain conditions.Citation16–19 This points to a genetic contribution to individual differences in chronic pain risk and/or severity, but the specific genetic architecture of CPSP remains incompletely understood. In addition, shared environmental factors are responsible for 7% to 10% variance in chronic pain development.Citation16 Similar to other chronic pain conditions, there is increasing evidence to show that genetic factors linked to CPSP riskCitation13,Citation20,Citation21 intricately interact with environmental factors to play a role in the transition of acute to chronic postsurgical pain.Citation22 Thus, in addition to genetics, epigenetic mechanisms have been a focus of study in development and maintenance of CPSP.
Though risk factors for CPSP and its related sequelae have been identified in clinical populations, the heterogeneity of patient demographics and surgical procedures, comorbidities, varying standards of care/pain definitions, and subjectivity of pain measures after surgery add complexity to clinical research.Citation23 Hence, preclinical models for CPSP are essential to understanding the pathological processes underlying CPSP and allow researchers to ask questions that could not be answered easily in the clinical setting. In this review, we discuss preclinical to clinical evidence for the role of genomics (genetics and epigenetics) in pediatric CPSP. We describe benefits and limitations of animal models used to study CPSP and discuss challenges of translational research. We also discuss epigenetic and genetic signatures in nociceptors and immune cells modulating neonatal nociceptive priming, an important concept leading to chronic pain transitions in children. We review current clinical studies in children describing genetic and epigenetic associations with CPSP and draw parallels with findings from adult genetic studies where there is a scarcity of pediatric evidence. Finally, we elaborate on integrative approaches of basic and clinical research, potential targets for novel therapeutic strategies in human subjects, and future areas of research.
To better understand the nuances of extrapolating adult findings to pediatric populations, it is important to understand the differences in physiology of the developing nociceptive system compared to adults. In adults, there is good evidence that amplification of neural signaling within the central nervous system leads to central sensitization, contributing to many prolonged chronic pain states.Citation24 However, an immature neonatal brain is not just a small adult brain. During brain development, a progressive reduction of intracellular chloride in neurons leading to an associated switch in gamma amino butyric acid (GABA) polarity (excitability and generation of depolarizing potentials in immature brains to hyperpolarization and inhibition) has been confirmed in a wide range of animal species.Citation25,Citation26 Also, it has been shown that up to postnatal day 21 (P21) in the rat, the rostroventral medulla of the brainstem exclusively facilitates spinal pain transmission but that after this age (P28 to adult), the influence of the rostroventral medulla shifts to biphasic facilitation and inhibition,Citation27 and this switch may be mediated by mu-opioid receptor pathways.Citation28 Although sensory neurons, including nociceptors, display age-related changes in functional makeup during early development,Citation5,Citation29 nociceptors can be functional by the 20th week of gestation. The peripheral sensory neurons in the dorsal root ganglia (DRG) overall appear to be fully developed by early childhood as external stimuli continue to shape their maturation.Citation29 However, interneuronal communications in the spinal cord are still developing at early ages. Hence, the premature newborn brain can poorly distinguish noxious and innocuous stimulation. Importantly, nociceptive reflexes and microglial reactions are strong at an early age, and repeated nociceptive stimuli (depending on age of initial insult) lead to irreversible changes that persist into adulthood, causing hyperalgesia, increasing risk for developing chronic pain, enhanced cortical activity to noxious stimulation, and considerable alterations in somatosensory and pain processing.Citation30,Citation31 We believe this brief prelude will highlight and provide a context for genomic evidence presented for pediatric CPSP as well as help readers understand relevant pediatric connections where adult findings are described in the article.Citation27,Citation28
Preclinical Models Relevant to Pediatric CPSP
Though there are many models that exist to study genomic/genetic/epigenetic factors contributing to postsurgical pain, only a subset of these are commonly applied to CPSP explicitly, and even fewer have been leveraged to investigate pediatric CPSP specifically. Refer to for a brief overview of preclinical surgical models relevant to pediatric CPSP genomic investigations. Detailed reviews on pain assessments in experimental models of neonatal and pediatric pain from early life sensitization have been previously published.Citation6,Citation32 Rodents (primarily mice and rats) are the most common animal model for pain genetics research, but there are several caveats to using these models for the study of pediatric CPSP. Mice are born at an earlier point of maturation compared with full-term birth in a human, equating roughly to the second postnatal week in rodents.Citation33 In addition, general maturational rates are not linearly correlated between rodents and humans; mice mature at ~150 times the rate of humans in the first month, and this ratio decreases to 25:1 after 6 months of age. As a result, if pediatric CPSP is defined clinically as pain lasting >3 months, this would correspond to ~14 hours in the first month of life for a mouse,Citation34 but most studies have used a much more protracted time frame (on the order of days to weeks depending on the specific surgical model) for measuring hypersensitivity after surgery/injury in adolescent mouse models, even in this early period of accelerated development. Quantification of chronic pain severity in these models is often accomplished using pain-eliciting stimuliCitation35–41 where severity of pain is associated with the degree of hypersensitivity exhibited or through pain measures such as alterations in gait and locomotor activity that more effectively mimic movement-evoked pain as seen during surgical recovery.Citation42
Figure 1. Diagrammatic representation of preclinical pain models, tests, and analyses used in genomic studies with relevance to pediatric chronic postsurgical pain phenotypes.
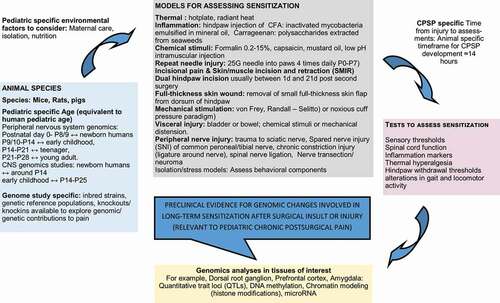
Preclinical models have proven useful for studying the effectiveness of common therapeutics for acute pain due to injury or inflammation (e.g., morphine, gabapentin, etoricoxib, celecoxib, indomethacin, naproxen) in the prevention and treatment of CPSP.Citation43,Citation44 However, pediatric studies are needed to allow translation of findings in adult to pediatric applications. For example, the development of anti-calcitonin gene-related peptide antibodies for the treatment of migraines in adults is currently being followed up by pediatric pharmacokinetic studiesCitation45 to determine whether dosing schedules based on weight or body surface area or hybrid models are optimal, because younger children have faster clearance and lower plasma concentrations when dosed based on weight and age.Citation46 In addition, safety, potential immunogenicity, and effects on pediatric physiology may be very different from those for adults. Despite the unknowns, which can only be resolved by long-term safety and efficacy trials in children, recommendations for its use in children with refractory migraine have been put forth by experts,Citation47 showing promise for potential translational success on CPSP therapeutics in children.Citation48 Nevertheless, animal models have limitations for translation. Behavioral responses to pain differ widely, with no clear-cut patterns between rats from the same strain purchased from different suppliers and different strains of mice, influencing both genetic association and interventional findings.Citation49,Citation50 Because pain is a biopsychosocial phenomenon, it is not amenable to assess the wholesome nature of this phenotype in animals, although certain models (acetic acid [0.9%] writhing test and manipulating social partner) have been used to simulate social environments. Despite interesting targets for therapies in animals (for example, neurokinin 1 antagonists), translation to human domains has been elusive.Citation51 Nevertheless, pain memory, an important risk predictor in pediatric acute to chronic pain transitions,Citation52 has been observed in animal studies showing long-term sensitivity following injury.Citation53 In addition, reverse translation, by first identifying variants associated with CPSP in human studies followed by mechanistic investigations in animal models, is suggested as a potentially improved approach to bridge the gap between benchside research and bedside applications.Citation54,Citation55
Preclinical Genetic Evidence in Chronic Postsurgical Pain
Findings from unbiased genome-wide approaches in animal models that recapitulate the tissue damage/injury aspects of surgery can provide insight into potential pediatric CPSP-relevant candidate genes. One such method, quantitative trait locus (QTL) mapping, has successfully identified multiple genomic loci in rodents where genotype is correlated with variation in the susceptibility to chronic pain; though these studies have not been conducted in juvenile mice, the data can be used to generate hypotheses for subsequent testing in pediatric CPSP. We could find no genomewide analysis conducted in animal models with the goal of identifying potential risk alleles or variants for pediatric-specific CPSP. However, two relevant QTLs, pain1 (mouse chromosome 15)Citation56 and pain2 (rat chromosome 2),Citation57 conducted in adult rodents have identified genomic loci associated with chronic pain in the neuroma model of sciatic nerve transection that shares similarities to surgical and traumatic amputations. Pain1 contains 155 genes, but using whole genome microarray expression analysis and bioinformatics, a single high-priority candidate, Cacng2, was identified. A Cacng2 hypomorphic mutant mouse confirmed the gene’s functional role in chronic pain susceptibility, and subsequent translational studies revealed human CACNG2 single-nucleotide polymorphisms (SNPs) predicted risk for CPSP in adult women.Citation57 A similar approach was used to identify and confirm a role for purinergic receptor P2rx7Citation58 in susceptibility for nerve injury–induced mechanical hypersensitivity. This provides strong support for uncovering the genetic basis for CPSP with genome-wide linkage mapping or similar preclinical tools. Animal models offer the opportunity to examine the role of specific candidate genes identified in clinical populations.Citation13 A direct example of this approach for CPSP comes from work by Tian et al.,Citation59 who sequenced 638 SNPs associated with 54 candidate pain-related genes in patients with CPSP and, as a result, identified brain-derived neurotrophic factor as a high-priority candidate gene. Knock-in mice harboring this specific brain-derived neurotrophic factor mutation were found to have decreased mechanical sensitivity corresponding to their human cohorts, indicating lower risk for CPSP. Though these methods are available and reliable, their application to pediatric CPSP has lagged behind their application to other forms of chronic pain.
Even with the paucity of unbiased whole-genome approaches being used in preclinical models of pediatric CPSP, future animal studies are critical to disentangling the individual differences involved in CPSP risk by offering (1) an enhanced level of precision for identifying the location, timing, and specific mechanisms by which individual genomic differences (genetic, or epigenetic) contribute to the pathology underlying pediatric CPSP and (2) a substrate for discovery of alternative therapeutics for treatment and prevention of pediatric CPSP. The fundamental genetic/epigenetic contributions to pediatric CPSP have yet to be identified, but the systematic control over environmental parameters in animal studies makes them ideal for this type of inquiry, and eventually these methods could be used to model the multiple clinical factors that likely contribute to CPSP in the clinical setting, including insufficient postoperative pain control,Citation60 presence of drains, postoperative infection,Citation61 and postponing the use of antineuropathic medication.Citation62–64
Preclinical Evidence for Epigenetic Mechanisms in Chronic Postsurgical Pain
Epigenetic modifications alter gene expression without altering the DNA sequence through processes including DNA methylation,Citation65 chromatin remodeling through histone modifications (methylation and acetylation), and noncoding RNAs (e.g., miRNAs)Citation66–72 that regulate gene expression.Citation67,Citation73 Prior work illustrates a number of specific alterations in epigenetic status induced in models of surgery-like injury, but, again, the application of these findings to the pediatric-equivalent in rodents is extremely limited. Nerve injury has been shown to induce global DNA hypomethylation in the DRG but global hypermethylation in the spinal cord and prefrontal cortex, pointing to the importance of tissue-specific changes in interpretation. To this end, Denk et al. previously proposed persistent, postinjury epigenetic alterations at microglial enhancers in spinal mechanisms underlying pain chronicity.Citation74 Chronic painful neuropathy induces persistent DNA hypomethylation in the prefrontal cortex and amygdalaCitation68 with a concomitant increase in of Synaptotagmin II (syt2) expression, which plays a role in synaptic vesicle docking and as a calcium sensor for fast neurotransmitter release.Citation75 These findings specifically point to an anatomical and epigenetic substrate for the emergence of psychological comorbidities of chronic pain, but their impact in the context of the immature brain in pediatric patients is unclear.Citation68,Citation76 Similarly, peripheral inflammation induces active DNA demethylation of the cbs gene promoter region in primary sensory afferents, resulting in increased production of hydrogen sulfide and increased pain.Citation77,Citation78 Other reports implicate differential methylation and hypoxia-inducible factor 1 signaling pathway gene expression in neuropathic pain severity in both rodent models and breast cancer survivors.Citation79 Relevant to CPSP, increased methylation of the mu- and kappa-opioid receptor promoters in DRG neurons following nerve injury provides a potential mechanism underlying the opioid resistance of neuropathic pain in preclinical and clinical populations.Citation80
Histone deacetylase (HDAC) levels increase in the spinal cord as a result of peripheral inflammation and nerve injury, suggesting a role in pain persistence and/or chronicity.Citation77,Citation81 In fact, inhibiting spinal HDAC activity attenuates nerve injury–induced hypersensitivity.Citation82 To this end, neuropathic pain reduces histone methylation, resulting in persistent dysregulation of the immune response to nerve injury.Citation83 Though these data are not specific to pediatric CPSP, they do shed light on potential therapeutic targets for the prevention of CPSP given the involvement of both inflammation and tissue injury in most surgical procedures and the sensitivity of the epigenome to transient alterations during the pediatric developmental stage.
In one of the only specific investigations of CPSP, a rat model of lingual nerve injury, a common occurrence during routine oral surgery or facial trauma/reconstruction, lingual nerve expression levels of multiple miRNAs predicted to regulate inflammatory and pain-related pathway genes were correlated with pain behavior. The relationships held true when miRNA expression in lingual neuromas was correlated with patient pain ratings.Citation84 miRNAs may contribute to alterations in sensory neuron excitability through their regulation of sodium channel (Nav) expression levels. The miRNAs miR-96Citation85 and miR-7aCitation86 exert regulatory control over Nav1.3 following nerve injury; the specific deletion of the miRNA processing enzyme Dicer in the DRG reduces expression of Nav1.7, 1.8, and 1.9 channels and attenuates inflammatory pain behaviors.Citation87 Relevant to the use of opioids for postoperative pain, the Let-7 group of miRNAs has been implicated in the development of morphine tolerance, offering a potential mechanism by which miRNAs could play a role in the opioid resistance of CPSP that affects both adult and pediatric patients.Citation88 Further functional studies examining the tissue-specific roles of all epigenetic modifications in the emergence of CPSP are needed, and this is particularly true for their role in pediatric CPSP.
Neonatal Nociceptive Priming: Epigenetic and Genetic Signatures in Nociceptors
A critical concern with children experiencing early life pain is how development of the nociceptive system is affected. Clinical and rodent data demonstrate that there are discrete time periods in which an aversive stimulus, such as an injury, results in altered development and long-lasting changes to the somatosensory system.Citation4,Citation89 Individuals who experience early life pain are at an increased risk of complications after an injury later in life, a phenomenon called neonatal nociceptive priming.Citation90,Citation91 Importantly, these effects are clinically relevant to neonates who undergo painful stimuli within the NICU. Hypersensitivity to tissue damage resulting from repeated heel sticks/procedures during clinical neonatal intensive care can persist long-term.Citation6 The specific role of different sensory neurons has been extensively studied in adult pain, but the role of specific sensory neurons in the onset of neonatal pain is not clearly understood.Citation92,Citation93 Reports have indicated that age is a key factor that modulates pain after peripheral nerve injury.Citation93,Citation94
The normal development of somatosensory and pain processing is dependent on the sensory information from skin, muscle, and joints, which relay information to the spinal cord during the first few postnatal weeks.Citation92,Citation95,Citation96 Primary sensory neurons of the DRG that respond to touch, pain, temperature, itch, etc.,Citation97 are chemically and functionally heterogenous.Citation97,Citation98 DRGs undergo many phenotypic changes during early postnatal development that are regulated by target-derived neurotropic factors (NTs).Citation96,Citation97,Citation99,Citation100 These factors exhibit temporal influence on developing primary afferents and alter the responses of neonatal sensory neurons to peripheral stimuli in response to injury.Citation29,Citation93,Citation96 A functional switch from mechanically sensitive, thermally insensitive C-fibers to polymodal C-fibers during postnatal developmentCitation29 coincides with the previously described neurochemical switch in growth factor responsiveness.Citation101 Thus, peripheral injury prior to or after this critical period results in distinct sensitization patterns in the DRG neurons. Unique pharmacological and behavioral responses to injuries exist between developing and adult subjects, and this is observed in both patients and animal models.Citation31,Citation92 Potential neonatal-specific analgesic properties and mechanisms of nociceptive signaling also lend credence to the presence of a “primed” nociceptive system that enhances the response to re-injury later in life.Citation92,Citation102
The genetic landscape of human and animals models is known to play important roles in the onset and perpetuation of chronic pain stemming from early life injury.Citation12,Citation13,Citation67,Citation103 Recent evidence demonstrates that neonatal mechanisms of nociception are distinct from those of adulta,Citation29,Citation104,Citation105 and early life injury has been shown to change patient sensitivity to peripheral stimuli in adulthood.Citation93,Citation106 When considering alterations in development, previous data indicate differences in chromatin accessibility between early stages of life and later developmental time points across different cell types.Citation107 Cellular activity can alter epigenetic signatures, and immunological data suggest that innate immune cells use the epigenome as a form of cellular memory.Citation108,Citation109 Further, animal models have identified alterations in neuronal function and differentiation through epigenetic modifications.Citation110,Citation111 However, the effect of injury and the direct impact this has on the nociceptive system is unknown. The complex interactions and genetic variation between patients such as SNPsCitation12,Citation13,Citation67,Citation112 and epigenetic modifications have gained attention in the onset of early pain.Citation12,Citation66,Citation67 However, the cell types, systems, and localization of neonatal nociceptive priming remain pertinent questions.Citation113 It will be necessary to determine the underlying factors that contribute to the unique vulnerability of the neonate, especially at the level of the sensory neuron in the form of a cellular “memory.” Hence, the definitive classification of primary sensory neurons at the single cell level over time under normal and pathological conditions will help identify genes involved in sensory neuron function and their role in neonatal priming. We are working to determine how subpopulations of sensory neurons are altered through development and the impact that early life injury has on the different subtypes at the functional and epigenetic levels. This type of analysis will be of critical importance to determine whether early life surgical incision drives chromatin accessibility modifications that contribute to neonatal nociceptive priming.
Neonatal Nociceptive Priming: Role of Macrophages
It is clear that within the spinal cord, both dorsal horn circuitry and microglia, the macrophages of the central nervous system, are critical for neonatal nociceptive priming.Citation91,Citation114,Citation115 However, evidence suggests that peripheral input through the primary afferents is also necessary for this.Citation105,Citation116 Nociceptive input is transmitted via primary afferent nociceptors and is modulated by the immune system.Citation117 Importantly, macrophages undergo robust developmental changes in early life and experience a critical period that overlaps with the vulnerable period of the somatosensory system.Citation118–121 Following infection or injury, a number of biological factors are released to the affected tissue, and macrophages begin to populate the area.Citation117 The pro- or anti-inflammatory profile and presence of macrophages have been linked to patient and animal outcomes following surgical injury during development.Citation122,Citation123 Together, these data suggest that activated macrophages in the neonate are unique and important in acute nociception as well as a long-term predisposition to chronic pain.
A unique feature of the peripheral immune system is its known ability to retain cellular memory. Though this “memory” is best attributed in the adaptive immune response, the innate immune system can also establish memory. In animals lacking an adaptive immune system, macrophages recognize pathogens to which they were previously exposedCitation109 through the unique pro- or anti-inflammatory microenvironment, signaling cascades, and epigenetic modifications.Citation108,Citation124 The microenvironment in the tissue creates a signature of cytokines, chemokines, and growth factors known as pathogen-associated molecular patterns and/or damage associated molecular patterns.Citation125 These are recognized by innate immune cells, including dendritic cells, natural killer cells, and macrophages, by pattern recognition receptors. The activation of these “lock and key” signals to receptors on macrophages induces intracellular signaling cascades altering transcription factors and the epigenetic landscape, which contributes to the formation of the immune memory.
Other molecules that directly alter the genome, such as HDACs, also drive epigenetic changes by modulating specific promotor regions to induce or inhibit pro- or anti-inflammatory responses from effector cells. Chromatin alterations include poised chromatin (e.g., H3K4me3 and H3K27me3), heterochromatin (e.g., H3K27me3 only), and active chromatin (e.g., H3K27ac and/or H3K4me3 only) or repressive chromatin (H3K9me2).Citation126,Citation127 Each of these modifications can induce long-lasting changes in gene expressionCitation128 and are specific to the pattern of stimulation.Citation109 It is important to note that the epigenome in early life is unique in macrophages,Citation129 necessary for tissue resident development,Citation130 and is required for monocyte transition into macrophages.Citation131
Macrophages have been found to become either “trained” or “tolerant” to certain stimuli. If trained, macrophages that are restimulated with a factor that they had previously encountered will display an increased pro-inflammatory response. Opposing this, macrophages that become tolerant to repeat stimuli have a reduced inflammatory response. The difference between these has been traced to differential epigenetic regulation on the promotors of effector genes. For example, stimulation to trained immunity can result in persistent active chromatin marks, whereas stimulation to tolerant immunity results in repressive marks.Citation124,Citation132 In either case, after cessation of the cellular response following the “first hit,” the cell resumes similar activity. It is not until a “second hit” that the priming effect within the cell is observed.Citation132 The factors that regulate this and the epigenetic landscape following different stimulations have been recently reviewed by Fanucchi et al.Citation133 Although it is clear that macrophages have distinct responses after restimulation, the timescale, developmental vulnerability, and effect after injury are less explored and warrant further investigation.
After a tissue breaking injury, including surgery, a number of biological and cellular systems initiate the injury response and facilitate repair of the damage.Citation134 The peripheral immune and nervous systems work together by sending signals to one another in a bidirectional pattern and to alter the local microenvironment.Citation135,Citation136 Previous data and our recent unpublished data indicate that the microenvironment after a neonatal surgical injury may be unique from that of the adult.Citation105,Citation129 Our work further demonstrates that macrophages are necessary for animals to display acute pain-like behaviors after an early life incision as well as chronic pain-like behaviors after a repeat injury later in life. The mechanisms that underlie macrophage involvement in maintaining memory of early life surgical injury may be similar to the mechanisms that underlie macrophage involvement after an infection and may be controlled by the epigenome. The unique properties of the neonatal macrophage and immune response may contribute to the vulnerable periods for both the peripheral immune system and nervous system. Because neonatal macrophages display a unique epigenetic landscape compared to adults,Citation129 these data indicate that pediatric surgery may drive macrophage modifications that are long-lasting and affect injury outcomes later in life.
Clinical Studies of CPSP in the Pediatric Population
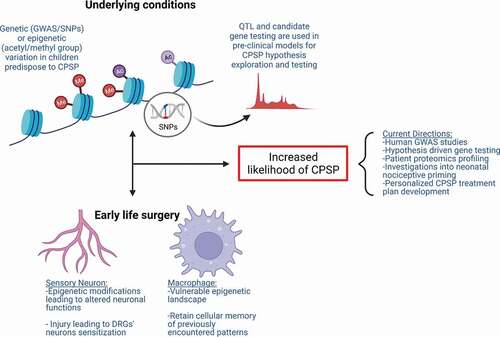
Pediatric clinical cohorts in CPSP genetic association studies are mostly small samples and thus findings need further scaling and validation. That said, the findings are mostly aligned with prior basic science knowledge, and novel systems biology–based approaches have been used to overcome size limitations. A schematic representation of the mechanisms involved in postinjury nociceptive priming from preclinical evidence is presented in .
Figure 2. Mechanisms contributing to increased susceptibility to CPSP. Underlying molecular mechanisms comprising genetic variations (i.e., SNPs) and epigenetic modifications (i.e., DNA or histone methylation and acetylation and miRNAs) contribute to individual differences in tissue-specific gene and protein expression in clinical association studies. Gene and protein expression differences can account for increased risk for altered neuronal excitability and sensitization. Alternative mechanisms involved in nociceptive priming are instigated following early life surgery. Tissue injury incites tissue-specific alterations (i.e., epigenetic modifications, gene expression changes) in cell types including sensory neurons and macrophages, which may be important in the formation and maintenance of neonatal nociceptive priming. Underlying conditions and early life surgery can independently contribute to increased susceptibility to CPSP and even act in a feedforward loop together, exacerbating CPSP.
Genetic Association Studies with CPSP
Recent systematic reviews describe CPSP–genetics associations.Citation137–139 In a comprehensive review of 21 CPSP gene association studies by Chidambaran et al., only one study included pediatric subjects (14–35 years) but the number of adolescents recruited was not stated.Citation140 They conducted a meta-analysis including six variants of five genes (COMT: rs4680 and rs6269, mu-1-opioid receptor/OPRM1: rs1799971, GTP cyclohydrolase 1/GCH1: rs3783641, potassium voltage-gated channel modifier subfamily S member 1/KCNS1: rs734784, tumor necrosis factor/TNFA: rs1800629),Citation141–149 but only rs734784 (A > G) of KCNS1 was found to marginally increase CPSP risk (additive genetic model; odds ratio = 1.511; 95% confidence interval [1–2.284]; P = 0.050). In another study, COMT rs4860 and μ-opioid receptor rs1799971 were not found to contribute to CPSP development after cesarean delivery.Citation150 Warner et al. conducted a GWAS meta-analysis and reported that a variant in protein kinase C alpha gene (PRKCA) gene was associated with neuropathic pain following total knee replacement,Citation151 but this was not replicated in other studies. Another GWAS Genome wide association studies in females posthysterectomy showed that rs118184265 at NAV3 was associated with CPSP in the replication cohort. Loci at cAMP response element-binding protein (CREB)‐regulated transcription co‐activator 3 gene (CRTC3) (rs117119665) associated with CREB-dependent transcription of genes and IQ motif containing GTPase‐activating protein 1 (IQGAP1) (rs1145324) involved in immune signaling were significantly associated with CPSP in a meta‐analysis in both the discovery and replication cohort.Citation152 However, the study was underpowered due to the small size of the discovery cohort. Heterogeneity in surgical cohorts, population structure, outcome definitions, unbalanced sex ratios, and the small cohort sizes are likely responsible for lack of consistent and replicable findings. For example, KCNS1 variant rs734784 A > G (Ile48Val) was associated with higher pain scores in patients with disc herniation and lumbar back pain, phantom limb and stump pain in amputees, preoperative sciatica pain, and experimental pain sensitivityCitation148 but not with long-term pain after breast cancer surgery,Citation153 raising the possibility that this variant might increase risk for neuropathic CPSP but not nonneuropathic pain.Citation154 summarizes the role of genes involved in variant CPSP association studies from the literature. It is unclear whether these findings will be replicated in pediatric cohorts. Although acute postsurgical pain and analgesic requirements are important predictors of CPSP in childrenCitation7 and genetic influences on both of these factors may play a role in CPSP, this is beyond the scope of this focused review on pediatric CPSP genomics. Detailed reviews on these aspects have been previously published.Citation155–157
Table 1. Literature-curated list of genes/variants associated with chronic postsurgical pain and their function.
Given difficulties in developing large genetic data banks with well-characterized CPSP phenotypes in children, leveraging systems biology may offer an alternative strategy to overcome sample size limitations.Citation174 Integrating genetic-level data with biologic processes can generate prioritized candidate gene lists. Chidambaran et al. demonstrated the utility of functional annotation–based prioritization and enrichment approaches to identify novel genes and unique/shared biological processes in acute and chronic postoperative pain.Citation175 Certain molecular mechanisms were elucidated to be common to acute and CPSP (e.g., CREB phosphorylation, ion channels, N-methyl-d-aspartate). Certain other genetic processes played a role in CPSP but not acute pain. These included immune/inflammatory (Toll-like receptor signaling, interferon gamma signaling, cytokines, mitogen-activated protein kinase/extracellular signal–regulated protein kinase signaling) and neurotransmitter-involved processes (purinergic, oxytocin, GABA, glutaminergic, catecholaminergic, dopaminergic). Despite the findings mostly being in adult studies, some of the pathways may be pertinent to pediatric populations, based on clinical and preclinical evidence. Several genes are common to immune, dopaminergic, serotoninergic, and catecholamine pathways (described in ). The latter three are also known to be involved in psychological disordersCitation176 implicated in the chronification of pain in children. For example, genes involved in dopaminergic neurotransmission (catechol-O-methyl transferase [COMT], GTP cyclohydrolase 1 [GCH1], and dopamine receptor [DRD2]) have different mechanisms.Citation177 GCH1 is involved in the production of BH4, a key molecule in the synthesis of dopamine, and variants (rs841) decrease GCH1 expression and are generally protective in chronic pain.Citation178 COMT is involved in degradation of dopamine and other catecholamines with key roles in chronic pain.Citation179 Its variants rs4680 and rs165774 decrease its enzymatic activity, increase catecholamine availability, and alter the signaling cascade. The dopaminergic receptors (D1-like receptor [D1LR] family [includes D1 and D5 receptors, which are stimulatory] and D2-like receptor [D2LR] family [consisting of D2, D3, and D4 receptors, which are inhibitory]) have opposite effects on nociceptive transmission. Variant rs6277 located in DRD2 decreases the stability of mRNA, thereby decreasing the expression of the D2 receptor, and increases CPSP risk.Citation180,Citation181 This pathway in modulation of nociception after surgery thus presents excellent targets for prevention and treatment of CPSP.Citation182–184
Because single variants account only for small effect sizes and different pathways play concomitant roles in CPSP development, one must consider the combined effect of several gene variants (polygenic risk) in CPSP.Citation17 Polygenic risk scores (PRSs)—the sum of weighted effects of different phenotype-associated alleles—have been shown to predict several complex conditions.Citation185–187 An atlas of PRS associations and putative causal relationships across the human phenome was reported, though it did not include CPSP as a phenotype.Citation188 Chidambaran et al. recently combined systems biology and penalized regression techniques to determine PRS, which improved prediction of CPSP risk compared to nongenetic models.Citation189 Another recent study determined a PRS that suggested significant overlap of genetics of CPSP with chronic widespread pain, rheumatoid arthritis, and sciatica (but not with chronic headache and migraine). They suggested that this overlap is potentially due to common mechanisms regulating neurological signaling (sodium channels) and inflammatory response.Citation190 Interestingly, this overlap was nullified in the replication cohort when subjects were randomly reassigned. Thus, further research is needed to enumerate polygenic risk for therapeutic targeting.Citation191,Citation192
Epigenetic Association with Clinical CPSP in Children
Epigenetic differences prior to surgery could serve as a risk factor for CPSP and tissue-specific epigenetic changes in response to a given surgery could serve as a separate risk factor.Citation193–203 As evidence of epigenetic regulation of CPSP risk, offspring of mothers fed a high methyl donor diet during the perinatal period exhibit increased acute pain (mechanical allodynia following skin incision),Citation204–206 highlighting the influence of DNA methylation patterns in susceptibility of injury-related pain. However, epigenetic association studies with CPSP are currently scarceCitation207 and present critical research gaps, especially in pediatrics. Using C-reactive protein as a marker, epigenome-wide association studies identified hypomethylated genes contributing to inflammatory processes in CPSP.Citation208 CpG methylation within tumor necrosis factor (TNF) gene promoter has been found to be a mechanism by which TNF alters risk for mild persistent breast pain in patients with breast cancer undergoing surgery.Citation153 DNA methylation at the promoter region of the mu-opioid receptor gene (OPRM1) that codes for mu-opioid receptor and important in opioid pain pathways has been studied.Citation209 DNA methylation at the promoter is a potent epigenetic repressor of gene transcriptionCitation210,Citation211 and is elevated in individuals addicted to opioids and heroin.Citation212,Citation213 In children undergoing spine fusion, blood DNA methylation in an active regulatory region of OPRM1 gene was associated with CPSP.Citation214 This region binds multiple transcription factors. It was postulated that inhibition of transcription factor binding by DNA methylation may decrease OPRM1 gene expression, leading to decreased opioid response and increased pain responses. In contrast, another study used machine learning methods to examine a potential association between the DNA methylation of two key players of glial/opioid intersection and persistent postoperative pain 3 years after breast cancer surgery.Citation215 Though their study supported a predictive utility of epigenetic testing using global DNA methylation, quantified at CpG sites located in the retrotransposon LINE1, they did not find that DNA methylation of two key genes of the glial–opioid interface (OPRM1 and Toll-like receptor TLR4) contributed to the persistent pain phenotype. Chidambaran et al. investigated whole blood DNA methylation profiles using epigenome-wide association studies to identify shared, enriched genomic pathways underlying CPSP and anxiety sensitivity Childhood Anxiety Sensitivity Index (CASI), recognized to increase CPSP risk.Citation7,Citation216 They identified 637 CPSP-associated and 2445 CASI-associated differentially DNA methylated positions (DMPs). The DMPs associated with both phenotypes enriched GABA receptor and dopamine-DARPP32 feedback in cyclic adenosine monophosphate signaling pathways. Using bioinformatic approaches, the authors elucidated target transcription factors and downstream modifying pathways regulating genes with DMP. Aligned with the GABA findings, rodent studies have identified preoperative anxiety-induced glucocorticoid signaling downregulated Npas4 (a neuronal PAS domain protein) leading to impaired spinal GABAergic system and ultimately contributing to postoperative hyperalgesia.Citation217 A schematic showing the presurgical genomic mechanisms that might increase risk for CPSP is depicted in .
Blood DNA methylation studies may identify CPSP biomarkers. Although environmental stressor changes are expected to be similar across tissues,Citation218 blood-based studies (for a neurological phenotype such as pain) could have limited mechanistic interpretation because DNA methylation is tissue specific. Although cell-free DNA (cfDNA) has not been studied in association with CPSP, reports of circulating cfDNA associations with inflammation and brain diseases such as schizophreniaCitation219,Citation220 point to potential use of cfDNA as a possible alternative to identify tissue-specific DNA methylation patterns.Citation221 Functional magnetic resonance imaging and spectroscopy could also be used to identify specific brain patterns and neurotransmitters associated with CPSP epigenetic findings.Citation222 However, because evidence does indicate a strong role for peripheral immune cells in CPSP development (see above), data could still play an important role in our understanding of the epigenetics of CPSP.
Postinjury and postsurgery epigenetic changes have not been studied in detail in vivo.Citation223 The few cross-sectional studies cannot capture dynamic epigenetic mechanisms, making it difficult to identify direction of causality.Citation224 Prospective longitudinal studies are needed to address reverse causation (epigenomes influenced by, rather than causal of, pain maintenance states). Within-subject studies will also be necessary to help control for potential confounders from associations of heritable SNPs with large DNA methylation–level differences near polymorphisms (cis effects) and associations of DNA methylation level differences with variants elsewhere in the human genome (trans effects).Citation225
Niculescu identified pain-related blood gene expression biomarkers for CPSP (MFAP3, GNG7, CNTN1, LY9, CCDC144B, and GBP1), some of which are targets of existing drugs.Citation226 There are plasma and cerebrospinal fluid biomarkers associated with pain,Citation227 but many of these remain unexplored in relation to CPSP.
MeQTLs: At the Intersection of Genetics and Epigenetics
Characterizing the complex relationship between genetic, epigenetic, and transcriptomic variation has the potential to increase understanding about the mechanisms underpinning CPSP phenotypes and how to influence the risk. Understanding gene–environment interactions underlying CPSP is an important area of research that is yet not well explored. One such mechanism includes methylation quantitative trait loci (meQTL), which are variants that influence DNA methylation at close or distant genomic loci. meQTLs were recently evaluated as mediators of genetic association with CPSP in a study in adolescents undergoing spine fusion.Citation228 Their rationale was based on the overlap of genetic variant and DNA methylation–enriched pathways associated with CPSP that they had previously reported on. This pilot study utilized causal inference tests to report that DNA methylation at 127 cytosine–guanine loci mediated association of 470 meQTLs with CPSP. They noted that several CpG–meQTL pairs were annotated to differentially methylated regions located at PARK16 locus on Chromosome 1, where CPSP risk meQTLs were associated with decreased DNA methylation at RAB7L1 and increased DNA methylation at PM20D1 genes. This region has previously been implicated in dopamine processing disorders of the nervous system.
Future Directions and Emerging Therapeutics and Interventions
Forward (bench to bedside) as well as backward (clinical to basic science) translation is needed to determine innovative targets and CPSP risk mitigation strategies. It is too early for tests based on newly discovered associations to provide stable estimates of genetic risk for CPSP. Although major findings are unlikely to be false positives, estimates based on combinations of current risk alleles need constant revision as new loci are found. In addition, CPSP may be too diverse a phenotype to have common genomic underpinnings—perhaps, study of endophenotypes and subgroups of patients having different characteristics based on biological pathways involved in the nature of pain (for example, predominantly nociceptive versus neuropathic), surgical nature (for example, musculoskeletal versus visceral), and socio-behavioral features will be a solution, as has been applied in developmental psychopathology.Citation229 Furthermore, inclusion of children and CPSP as a phenotype (especially now that is a recognized International Classification of Diseases, 11th Revision entity)1 within large-scale genetic studies (for example, the UK Biobank registry)Citation188 would allow genome-wide approaches to pediatric CPSP. Thus, we remain optimistic that in the future, genetics combined with other biomarkers could preoperatively stratify CPSP risk, guiding prevention and treatment. Though some gene association studies also investigated gene–gene,Citation142 gene–sex,Citation230 and gene–psychological factor interactions,Citation231 research of such interactions, including gene–epigenetic interactions,Citation211 is still in its infancy, and further research is needed to understand acute to chronic postsurgical pain transition, especially in children.
Several promising emerging therapeutics targeting genes and proteins first identified in animal models and involved in the transition from acute to chronic pain have been detailed previously.Citation134,Citation193,Citation202,Citation232–236 Gene editingCitation237 and the development of novel chemical decoysCitation238,Citation239 that target the neurobiological substrates of chronic pain offer the potential for precision pain management strategies based on manipulating this genetic context to effectively protect patients from CPSP without the negative side effects of opioids. Though a new CPSP treatment option has been slow to emerge, understanding and targeting genes, gene expression, and the processes that regulate expression represent a logical next step in developing precision pain management for CPSP.
Epigenetic biomarkers are being developed for screening in some areas like cancer. They are also being used to develop therapeutic targets. Sun et al. found that DNA methyltranferase (DNMT) inhibitor 5-Aza-2ʹ-deoxycytidine significantly reduced incision-induced mechanical allodynia and thermal sensitivity.Citation240 Although six epigenetic drugs are approved for use in the United States (many more under development), their nonspecific effects are a significant drawback (see reviewsCitation241,Citation242). In addition to generalized epigenetic targeting approaches, gene-specific epigenetic targeting is becoming a possibility through recently developed genome editing technology (e.g., demethylation of specific CpGs in human cells using fusions of engineered transcription activator–like effector repeat arrays, TET1 hydroxylase catalytic domain) that can effectively target and demethylate individual genes in vitro.Citation243 In addition, Cas9 systems offer novel individual gene targeted approaches.Citation244 Interestingly, the beneficial effects of lifestyle modifications (e.g., exercise) on mechanical and thermal hypersensitivity after sciatic nerve injuryCitation245 are partially mediated by decreased HDAC activity and increased acetylation of histones in the spinal cord,Citation246 pointing to the potential use of nonpharmacologic strategies targeting the epigenome in the management of CPSP.
Pharmacogenomic profiles are also being generated for individual patients in order to develop better pain management strategies.Citation247,Citation248 For example, research on the mu-opioid receptor has depicted several polymorphisms that could lead to a tailored targeting of an identified SNP.Citation247 Similarly, there have been some studies using proteomics to study different types of pain,Citation249,Citation250 such as widespread musculoskeletal pain,Citation251 abdominal pain,Citation252 and low back pain.Citation253 Modifying the existing drugs to target these proteins’ functionality may achieve the goal of treating CPSP, but proteomics profiling of pediatric populations would be a required first step to determine the utility of this strategy.
Conclusion
There is much work to be done to understand pain-related genomics and DNA methylation changes, the crosstalk between modifiable environmental factors and pain, optimal times to intervene to prevent acute to chronic pain transitions, and identification of optimal pathways to target therapeutically. Future treatment may include epigenetically programmed drugsCitation254 or simple modifications to preoperative regimens, including nutrition,Citation255 activity, mindfulness, or behavioral therapy,Citation256–258 to prevent persistence of pain after injury or surgery. Distinct cellular interactions must also be taken into consideration in order to enhance translational potential. Clear evidence suggests a role for both neurons and immune cells (among others) in the epigenetic regulation of CPSP. Changing bidirectional communication between neurons and immune cells is essential for proper transduction of sensory stimuli over the life span and should therefore be contemplated when developing future treatments for CPSP in children.
Acknowledgment
The authors thank Maria Ashton MS, RPH, MBA, for providing writing assistance, editing, and proofreading.
Disclosure Statement
The authors have no conflict of interest to report.
Additional information
Funding
References
- Schug SA, Lavand’homme P, Barke A, Korwisi B, Rief W, Treede R-D. The IASP classification of chronic pain for ICD-11: chronic postsurgical or posttraumatic pain. Pain. 2019 Jan;160(1):45–52. doi:https://doi.org/10.1097/j.pain.0000000000001413.
- Rabbitts JA, Fisher E, Rosenbloom BN, Palermo TM. Prevalence and Predictors of chronic postsurgical pain in children: a systematic review and meta-analysis. J Pain. 2017 Jun;18(6):605–14. doi:https://doi.org/10.1016/j.jpain.2017.03.007.
- Peters JWB, Schouw R, Anand KJS, van Dijk M, Duivenvoorden HJ, Tibboel D. Does neonatal surgery lead to increased pain sensitivity in later childhood? Pain. 2005 Apr;114(3):444–54. doi:https://doi.org/10.1016/j.pain.2005.01.014.
- Hermann C, Hohmeister J, Demirakca S, Zohsel K, Flor H. Long-term alteration of pain sensitivity in school-aged children with early pain experiences. Pain. 2006 Dec 5;125(3):278–85. doi:https://doi.org/10.1016/j.pain.2006.08.026.
- Fitzgerald M, Millard C, McIntosh N. Cutaneous hypersensitivity following peripheral tissue damage in newborn infants and its reversal with topical anaesthesia. Pain. 1989;39(1):31–36. doi:https://doi.org/10.1016/0304-3959(89)90172-3.
- Williams MD, Lascelles BDX. Early neonatal pain-a review of clinical and experimental implications on painful conditions later in life. Front Pediatr. 2020;8:30. doi:https://doi.org/10.3389/fped.2020.00030.
- Chidambaran V, Ding L, Moore DL, Spruance K, Cudilo EM, Pilipenko V, Hossain M, Sturm P, Kashikar-Zuck S, Martin LJ, et al. Predicting the pain continuum after adolescent idiopathic scoliosis surgery: a prospective cohort study. Eur J Pain. 2017 Aug;21(7):1252–65. doi:https://doi.org/10.1002/ejp.1025.
- Batoz H, Semjen F, Bordes-Demolis M, Bénard A, Nouette-Gaulain K. Chronic postsurgical pain in children: prevalence and risk factors. A prospective observational study. BJA: British Journal of Anaesthesia. 2016;117(4):489–96. doi:https://doi.org/10.1093/bja/aew260.
- Page MG, Stinson J, Campbell F, Isaac L, Katz J. Identification of pain-related psychological risk factors for the development and maintenance of pediatric chronic postsurgical pain. J Pain Res. 2013;6:167–80. doi:https://doi.org/10.2147/JPR.S40846.
- Page MG, Campbell F, Isaac L, Stinson J, Katz J. Parental risk factors for the development of pediatric acute and chronic postsurgical pain: a longitudinal study. J Pain Res. 2013;6:727–41. doi:https://doi.org/10.2147/JPR.S51055.
- Rabbitts JA, Palermo TM, Lang EA. A conceptual model of biopsychosocial mechanisms of transition from acute to chronic postsurgical pain in children and adolescents. J Pain Res. 2020;13:3071–80. doi:https://doi.org/10.2147/JPR.S239320.
- Young EE, Lariviere WR, Belfer I. Genetic basis of pain variability: recent advances. J Med Genet. 2012 Jan;49(1):1–9. doi:https://doi.org/10.1136/jmedgenet-2011-100386.
- Clarke H, Katz J, Flor H, Rietschel M, Diehl SR, Seltzer Z. Genetics of chronic post-surgical pain: a crucial step toward personal pain medicine. Can J Anaesth. 2015 Mar;62(3):294–303. doi:https://doi.org/10.1007/s12630-014-0287-6.
- Diatchenko L, Nackley AG, Tchivileva IE, Shabalina SA, Maixner W. Genetic architecture of human pain perception. Trends Genet. 2007 Dec;23(12):605–13. doi:https://doi.org/10.1016/j.tig.2007.09.004.
- Angst MS, Phillips NG, Drover DR, Tingle M, Ray A, Swan GE, Lazzeroni LC, Clark DJ. Pain sensitivity and opioid analgesia: a pharmacogenomic twin study. Pain. 2012 Jul;153(7):1397–409. doi:https://doi.org/10.1016/j.pain.2012.02.022.
- Vehof J, Zavos HM, Lachance G, Hammond CJ, Williams FM. Shared genetic factors underlie chronic pain syndromes. Pain. 2014 Aug;155(8):1562–68. doi:https://doi.org/10.1016/j.pain.2014.05.002.
- Diatchenko L, Nackley AG, Slade GD, Fillingim RB, Maixner W. Idiopathic pain disorders–pathways of vulnerability. Pain. 2006 Aug;123(3):226–30. doi:https://doi.org/10.1016/j.pain.2006.04.015.
- Hocking LJ, Generation S, Morris AD, Dominiczak AF, Porteous DJ, Smith BH. Heritability of chronic pain in 2195 extended families. Eur J Pain. 2012 Aug;16(7):1053–63. doi:https://doi.org/10.1002/j.1532-2149.2011.00095.x.
- Peters MJ, Broer L, Willemen HL, Eiriksdottir G, Hocking LJ, Holliday KL, Horan MA, Meulenbelt I, Neogi T, Popham M, et al. Genome-wide association study meta-analysis of chronic widespread pain: evidence for involvement of the 5p15.2 region. Ann Rheum Dis. 2013 Mar;72(3):427–36. doi:https://doi.org/10.1136/annrheumdis-2012-201742.
- James SK. Chronic postsurgical pain: is there a possible genetic link? British Journal of Pain 1. 2017;11(4):178–185.
- Katz J, Seltzer Z. Transition from acute to chronic postsurgical pain: risk factors and protective factors. Expert Rev Neurother. 2009 May;9(5):723–44. doi:https://doi.org/10.1586/ern.09.20.
- Campbell P, Jordan KP, Smith BH, Scotland G, Dunn KM. Chronic pain in families: a cross-sectional study of shared social, behavioural, and environmental influences. Pain. 2018;159(1):41–47. doi:https://doi.org/10.1097/j.pain.0000000000001062.
- Meissner W, Coluzzi F, Fletcher D, Huygen F, Morlion B, Neugebauer E, Pérez AM, Pergolizzi J. Improving the management of post-operative acute pain: priorities for change. Curr Med Res Opin. 2015 Nov;31(11):2131–43. doi:https://doi.org/10.1185/03007995.2015.1092122.
- Woolf CJ. Pain amplification—A perspective on the how, why, when, and where of central sensitization. J Appl Biobehav Res. 2018;23(2):e12124. doi:https://doi.org/10.1111/jabr.12124.
- Ben-Ari Y, Gaiarsa J-L, Tyzio R, Khazipov R. GABA: a Pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. 2007;87(4):1215–84. doi:https://doi.org/10.1152/physrev.00017.2006.
- Ben-Ari Y, Woodin M, Sernagor E, Cancedda L, Vinay L, Rivera C, Legendre P, Luhmann HJ, Bordey A, Wenner P, et al. 2012 August 28. Refuting the challenges of the developmental shift of polarity of GABA actions: GABA more exciting than ever! Hypothesis and Theory. Front Cell Neurosci.6(35). doi:https://doi.org/10.3389/fncel.2012.00035.
- Hathway GJ, Koch S, Low L, Fitzgerald M. The changing balance of brainstem-spinal cord modulation of pain processing over the first weeks of rat postnatal life. J Physiol. 2009 Jun 15;587(Pt 12):2927–35. doi:https://doi.org/10.1113/jphysiol.2008.168013.
- Hathway GJ, Vega-Avelaira D, Fitzgerald M. A critical period in the supraspinal control of pain: opioid-dependent changes in brainstem rostroventral medulla function in preadolescence. Pain. 2012 Apr;153(4):775–83. doi:https://doi.org/10.1016/j.pain.2011.11.011.
- Jankowski MP, Ross JL, Weber JD, Lee FB, Shank AT, Hudgins RC. Age-dependent sensitization of cutaneous nociceptors during developmental inflammation. Mol Pain. 2014 Jun 7;10:34. doi:https://doi.org/10.1186/1744-8069-10-34.
- Hohmeister J, Demirakca S, Zohsel K, Flor H, Hermann C. Responses to pain in school-aged children with experience in a neonatal intensive care unit: cognitive aspects and maternal influences. Eur J Pain. 2009 Jan;13(1):94–101. doi:https://doi.org/10.1016/j.ejpain.2008.03.004.
- Walker SM, Tochiki KK, Fitzgerald M. Hindpaw incision in early life increases the hyperalgesic response to repeat surgical injury: critical period and dependence on initial afferent activity. Pain. 2009 Dec 15;147(1–3):99–106. doi:https://doi.org/10.1016/j.pain.2009.08.017.
- Walker SM. Biological and neurodevelopmental implications of neonatal pain. Clin Perinatol. 2013 Sep;40(3):471–91. doi:https://doi.org/10.1016/j.clp.2013.05.002.
- Clancy B, Silva-Filho M, Friedlander MJ. Structure and projections of white matter neurons in the postnatal rat visual cortex. J Comp Neurol. 2001 May 28;434(2):233–52. doi:https://doi.org/10.1002/cne.1174.
- Miller RA, Harrison DE, Astle CM, Floyd RA, Flurkey K, Hensley KL, Javors MA, Leeuwenburgh C, Nelson JF, Ongini E, et al. An aging interventions testing program: study design and interim report. Aging Cell. 2007 Aug;6(4):565–75. doi:https://doi.org/10.1111/j.1474-9726.2007.00311.x.
- Brennan TJ, Vandermeulen EP, Gebhart GF. Characterization of a rat model of incisional pain. Pain. 1996 Mar;64(3):493–502. doi:https://doi.org/10.1016/0304-3959(95)01441-1.
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988 Jan;32(1):77–88. doi:https://doi.org/10.1016/0304-3959(88)90026-7.
- Brennan TJ. Pathophysiology of postoperative pain. Pain. 2011 Mar;152(3 Suppl):S33–S40. doi:https://doi.org/10.1016/j.pain.2010.11.005.
- Chapman CR, Vierck CJ. The transition of acute postoperative pain to chronic pain: an integrative overview of research on mechanisms. J Pain. 2017 Apr;18(4):359 e1–359 e38. doi:https://doi.org/10.1016/j.jpain.2016.11.004.
- Pogatzki EM, Raja SN. A mouse model of incisional pain. Anesthesiology. 2003 Oct;99(4):1023–27. doi:https://doi.org/10.1097/00000542-200310000-00041.
- Cowie AM, Stucky CL. A mouse model of postoperative pain. Bio Protoc. 2019 Jan 20;9(2). doi:https://doi.org/10.21769/BioProtoc.3140.
- Cummins TM, Kucharczyk MM, Graven-Nielsen T, Bannister K. Activation of the descending pain modulatory system using cuff pressure algometry: back translation from man to rat. Eur J Pain. 2020 Aug;24(7):1330–38. doi:https://doi.org/10.1002/ejp.1580.
- Tappe-Theodor A, King T, Morgan MM. Pros and cons of clinically relevant methods to assess pain in rodents. Neurosci Biobehav Rev. 2019 May;100:335–43. doi:https://doi.org/10.1016/j.neubiorev.2019.03.009.
- Whiteside GT, Harrison J, Boulet J, Mark L, Pearson M, Gottshall S, Walker K. Pharmacological characterisation of a rat model of incisional pain. Br J Pharmacol. 2004 Jan;141(1):85–91. doi:https://doi.org/10.1038/sj.bjp.0705568.
- Yang J, Yuan F, Ye G, Wang Y-J, Wu C, Wang J, Li X-Y, Feng Z. Skin/muscle incision and retraction induces evoked and spontaneous pain in mice. Pain Res Manag. 2019;2019:6528528. doi:https://doi.org/10.1155/2019/6528528.
- Zheng S, Gaitonde P, Andrew MA, Gibbs MA, Lesko LJ, Schmidt S. Model-based assessment of dosing strategies in children for monoclonal antibodies exhibiting target-mediated drug disposition. CPT: Pharmacometrics & Systems Pharmacology. 2014 Oct 1;3(10):e138. doi:https://doi.org/10.1038/psp.2014.38.
- Malik P, Edginton A. Pediatric physiology in relation to the pharmacokinetics of monoclonal antibodies. Expert Opin Drug Metab Toxicol. 2018 Jun;14(6):585–99. doi:https://doi.org/10.1080/17425255.2018.1482278.
- Szperka CL, VanderPluym J, Orr SL, Oakley CB, Qubty W, Patniyot I, Lagman-Bartolome AM, Morris C, Gautreaux J, Victorio MC, et al. Recommendations on the use of anti-cgrp monoclonal antibodies in children and adolescents. Headache. 2018;58(10):1658–69. doi:https://doi.org/10.1111/head.13414.
- Edvinsson L, Haanes KA, Warfvinge K, Krause DN. CGRP as the target of new migraine therapies - successful translation from bench to clinic. Nat Rev Neurol. 2018 Jun;14(6):338–50. doi:https://doi.org/10.1038/s41582-018-0003-1.
- Mogil JS, Wilson SG, Bon K, Eun Lee S, Chung K, Raber P, Pieper JO, Hain HS, Belknap JK, Hubert L, et al. Heritability of nociception I: responses of 11 inbred mouse strains on 12 measures of nociception. Pain. 1999 Mar;80(1–2):67–82. doi:https://doi.org/10.1016/s0304-3959(98)00197-3.
- Akhtar A. The flaws and human harms of animal experimentation. Cambridge Quarterly of Healthcare Ethics. 2015;24(4):407–19. doi:https://doi.org/10.1017/S0963180115000079.
- Borsook D, Upadhyay J, Klimas M, Schwarz AJ, Coimbra A, Baumgartner R, George E, Potter WZ, Large T, Bleakman D, et al. Decision-making using fMRI in clinical drug development: revisiting NK-1 receptor antagonists for pain. Drug Discov Today. 2012 Sep;17(17–18):964–73. doi:https://doi.org/10.1016/j.drudis.2012.05.004.
- Noel M, McMurtry CM, Pavlova M, Taddio A. Brief clinical report: a systematic review and meta-analysis of pain memory-reframing interventions for children’s needle procedures. Pain Pract. 2018 Jan;18(1):123–29. doi:https://doi.org/10.1111/papr.12572.
- Bhutta AT, Rovnaghi C, Simpson PM, Gossett JM, Scalzo FM, Anand KJ. Interactions of inflammatory pain and morphine in infant rats: long-term behavioral effects. Physiol Behav. 2001 May;73(1–2):51–58. doi:https://doi.org/10.1016/s0031-9384(01)00432-2.
- Martin LJ, Smith SB, Khoutorsky A, Magnussen CA, Samoshkin A, Sorge RE, Cho C, Yosefpour N, Sivaselvachandran S, Tohyama S, et al. Epiregulin and EGFR interactions are involved in pain processing. J Clin Invest. 2017 Sep 1;127(9):3353–66. doi:https://doi.org/10.1172/jci87406.
- Cho C, Deol HK, Martin LJ. Bridging the translational divide in pain research: biological, psychological and social considerations. Front Pharmacol. 2021;12:603186. doi:https://doi.org/10.3389/fphar.2021.603186.
- Devor M, Gilad A, Arbilly M, Yakir B, Raber P, Pisanté A, Darvasi A. pain1: a neuropathic pain QTL on mouse chromosome 15 in a C3HxC58 backcross. Pain. 2005 Aug;116(3):289–93. doi:https://doi.org/10.1016/j.pain.2005.04.023.
- Nissenbaum J, Shpigler H, Pisante A, delCanho S, Minert A, Seltzer Z, Devor M, Darvasi A. pain2: a neuropathic pain QTL identified on rat chromosome 2. Pain. 2008 Mar;135(1–2):92–97. doi:https://doi.org/10.1016/j.pain.2007.05.006.
- Sorge RE, Trang T, Dorfman R, Smith SB, Beggs S, Ritchie J, Austin J-S, Zaykin DV, Meulen HV, Costigan M, et al. Genetically determined P2X7 receptor pore formation regulates variability in chronic pain sensitivity. Nat Med. 2012;18(4):595–99. doi:https://doi.org/10.1038/nm.2710.
- Tian Y, Liu X, Jia M, Yu H, Lichtner P, Shi Y, Meng Z, Kou S, Ho IHT, Jia B, et al. Targeted genotyping identifies susceptibility locus in brain-derived neurotrophic factor gene for chronic postsurgical pain. Anesthesiology. 2018 Mar;128(3):587–97. doi:https://doi.org/10.1097/ALN.0000000000001977.
- Katz J, Jackson M, Kavanagh BP, Sandler AN. Acute pain after thoracic surgery predicts long-term post-thoracotomy pain. Clin J Pain. 1996 Mar;12(1):50–55. doi:https://doi.org/10.1097/00002508-199603000-00009.
- Lirk P, Fiegl H, Weber NC, Hollmann MW. Epigenetics in the perioperative period. Br J Pharmacol. 2015 Jun;172(11):2748–55. doi:https://doi.org/10.1111/bph.12865.
- Rosenbloom BN, Page MG, Isaac L, Campbell F, Stinson JN, Wright JG, Katz J. pediatric chronic postsurgical pain and functional disability: a prospective study of risk factors up to one year after major surgery. J Pain Res. 2019;12:3079–98. doi:https://doi.org/10.2147/JPR.S210594.
- Williams G, Howard RF, Liossi C. Persistent postsurgical pain in children and young people: prediction, prevention, and management. Pain Rep. 2017 Sep;2(5):e616. doi:https://doi.org/10.1097/PR9.0000000000000616.
- Kinney MA, Jacob AK, Passe MA, Mantilla CB. increased risk of postthoracotomy pain syndrome in patients with prolonged hospitalization and increased postoperative opioid use. Pain Res Treat. 2016;2016:7945145. doi:https://doi.org/10.1155/2016/7945145.
- Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005 Aug;6(8):597–610. doi:https://doi.org/10.1038/nrg1655.
- Doehring A, Oertel BG, Sittl R, Lötsch J. Chronic opioid use is associated with increased DNA methylation correlating with increased clinical pain. Pain. 2013 Jan;154(1):15–23. doi:https://doi.org/10.1016/j.pain.2012.06.011.
- James SK. Chronic postsurgical pain: is there a possible genetic link? Br J Pain. 2017 Nov;11(4):178–85. doi:https://doi.org/10.1177/2049463717723222.
- Tajerian M, Alvarado S, Millecamps M, Vachon P, Crosby C, Bushnell MC, Szyf M, Stone LS. Peripheral nerve injury is associated with chronic, reversible changes in global DNA methylation in the mouse prefrontal cortex. PLoS One. 2013;8(1):e55259. doi:https://doi.org/10.1371/journal.pone.0055259.
- Shi G, Shi J, Liu K, Liu N, Wang Y, Fu Z, Ding J, Jia L, Yuan W. Increased miR-195 aggravates neuropathic pain by inhibiting autophagy following peripheral nerve injury. Glia. 2013 Apr;61(4):504–12. doi:https://doi.org/10.1002/glia.22451.
- Bird A. The dinucleotide CG as a genomic signalling module. J Mol Biol. 2011 May 27;409(1):47–53. doi:https://doi.org/10.1016/j.jmb.2011.01.056.
- Varela MA, Roberts TC, Wood MJ. Epigenetics and ncRNAs in brain function and disease: mechanisms and prospects for therapy. Neurotherapeutics. 2013 Oct;10(4):621–31. doi:https://doi.org/10.1007/s13311-013-0212-7.
- Abel T, Zukin RS. Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr Opin Pharmacol. 2008 Feb;8(1):57–64. doi:https://doi.org/10.1016/j.coph.2007.12.002.
- Anacker C, O’Donnell KJ, Meaney MJ. Early life adversity and the epigenetic programming of hypothalamic-pituitary-adrenal function. Dialogues Clin Neurosci. 2014 Sep;16(3):321–33. doi:https://doi.org/10.31887/DCNS.2014.16.3/canacker.
- Denk F, Crow M, Didangelos A, Lopes DM, McMahon SB. Persistent alterations in microglial enhancers in a model of chronic pain. Cell Rep. 2016 May 24;15(8):1771–81. doi:https://doi.org/10.1016/j.celrep.2016.04.063.
- Alvarado S, Tajerian M, Suderman M, Machnes Z, Pierfelice S, Millecamps M, Stone LS, Szyf M. An epigenetic hypothesis for the genomic memory of pain. Front Cell Neurosci. 2015;9:88–88. doi:https://doi.org/10.3389/fncel.2015.00088.
- Alvarado S, Tajerian M, Millecamps M, Suderman M, Stone LS, Szyf M. Peripheral nerve injury is accompanied by chronic transcriptome-wide changes in the mouse prefrontal cortex. Mol Pain. 2013 Apr 18;9:21. doi:https://doi.org/10.1186/1744-8069-9-21.
- Bai G, Wei D, Zou S, Ren K, Dubner R. Inhibition of class II histone deacetylases in the spinal cord attenuates inflammatory hyperalgesia. Mol Pain. 2010 Sep 7;6:51. doi:https://doi.org/10.1186/1744-8069-6-51.
- Qi F, Zhou Y, Xiao Y, Tao J, Gu J, Jiang X, Xu G-Y. Promoter demethylation of cystathionine-beta-synthetase gene contributes to inflammatory pain in rats. Pain. 2013 Jan;154(1):34–45. doi:https://doi.org/10.1016/j.pain.2012.07.031.
- Kober KM, Lee MC, Olshen A, et al. Differential methylation and expression of genes in the hypoxia-inducible factor 1 signaling pathway are associated with paclitaxel-induced peripheral neuropathy in breast cancer survivors and with preclinical models of chemotherapy-induced neuropathic pain. Mol Pain Jan-Dec. 2020;16:1744806920936502. doi:https://doi.org/10.1177/1744806920936502.
- Sun L, Zhao JY, Gu X, Liang L, Wu S, Mo K, Feng J, Guo W, Zhang J, Bekker A, et al. Nerve injury-induced epigenetic silencing of opioid receptors controlled by DNMT3a in primary afferent neurons. Pain. 2017 Jun;158(6):1153–65. doi:https://doi.org/10.1097/j.pain.0000000000000894.
- Zhang Z, Cai Y-Q, Zou F, Bie B, Pan ZZ. Epigenetic suppression of GAD65 expression mediates persistent pain. Nat Med. 2011;17(11):1448–55. doi:https://doi.org/10.1038/nm.2442.
- Denk F, Huang W, Sidders B, Bithell A, Crow M, Grist J, Sharma S, Ziemek D, Rice ASC, Buckley NJ, et al. HDAC inhibitors attenuate the development of hypersensitivity in models of neuropathic pain. Pain. 2013 Sep;154(9):1668–79. doi:https://doi.org/10.1016/j.pain.2013.05.021.
- Imai S, Ikegami D, Yamashita A, Shimizu T, Narita M, Niikura K, Furuya M, Kobayashi Y, Miyashita K, Okutsu D, et al. Epigenetic transcriptional activation of monocyte chemotactic protein 3 contributes to long-lasting neuropathic pain. Brain. 2013 Mar;136(Pt 3):828–43. doi:https://doi.org/10.1093/brain/aws330.
- Tavares-Ferreira D, Lawless N, Bird EV, Atkins S, Collier D, Sher E, Malki K, Lambert DW, Boissonade FM. Correlation of miRNA expression with intensity of neuropathic pain in man. Mol Pain Jan-Dec. 2019;15:1744806919860323. doi:https://doi.org/10.1177/1744806919860323.
- Chen HP, Zhou W, Kang LM, et al. Intrathecal miR-96 inhibits Nav1.3 expression and alleviates neuropathic pain in rat following chronic construction injury. Neurochem Res. 2014 Jan;39(1):76–83. doi:https://doi.org/10.1007/s11064-013-1192-z.
- Sakai A, Saitow F, Miyake N, Miyake K, Shimada T, Suzuki H. miR-7a alleviates the maintenance of neuropathic pain through regulation of neuronal excitability. Brain. 2013 Sep;136(Pt 9):2738–50. doi:https://doi.org/10.1093/brain/awt191.
- Zhao J, Lee MC, Momin A, Cendan C-M, Shepherd ST, Baker MD, Asante C, Bee L, Bethry A, Perkins JR, et al. Small RNAs control sodium channel expression, nociceptor excitability, and pain thresholds. J Neurosci. 2010 Aug 11;30(32):10860–71. doi:https://doi.org/10.1523/JNEUROSCI.1980-10.2010.
- He XY, Chen JX, Zhang Z, Li CL, Peng QL, Peng HM. The let-7a microRNA protects from growth of lung carcinoma by suppression of k-Ras and c-Myc in nude mice. J Cancer Res Clin Oncol. 2010 Jul;136(7):1023–28. doi:https://doi.org/10.1007/s00432-009-0747-5.
- Reynolds ML, Fitzgerald M. Long-term sensory hyperinnervation following neonatal skin wounds. J Comp Neurol. 1995 Aug 7;358(4):487–98. doi:https://doi.org/10.1002/cne.903580403.
- Walker SM. Early life pain—effects in the adult. Current Opinion in Physiology. 2019 Oct 1;11:16–24. doi:https://doi.org/10.1016/j.cophys.2019.04.011.
- Beggs S, Currie G, Salter MW, Fitzgerald M, Walker SM. Priming of adult pain responses by neonatal pain experience: maintenance by central neuroimmune activity. Brain. 2012 Feb;135(Pt 2):404–17. doi:https://doi.org/10.1093/brain/awr288.
- Fitzgerald M, Walker SM. Infant pain management: a developmental neurobiological approach. Nat Clin Pract Neurol. 2009 Jan;5(1):35–50. doi:https://doi.org/10.1038/ncpneuro0984.
- Fitzgerald M, McKelvey R. Nerve injury and neuropathic pain - A question of age. Exp Neurol. 2016;275(Pt 2):296–302. doi:https://doi.org/10.1016/j.expneurol.2015.07.013.
- Verriotis M, Chang P, Fitzgerald M, Fabrizi L. The development of the nociceptive brain. Neuroscience. 2016 Dec 3;338:207–19. doi:https://doi.org/10.1016/j.neuroscience.2016.07.026.
- La Hausse De Lalouviere L, Morice O, Fitzgerald M. Altered sensory innervation and pain hypersensitivity in a model of young painful arthritic joints: short- and long-term effects. Inflamm Res. 2021 Apr;70(4):483–93. doi:https://doi.org/10.1007/s00011-021-01450-5.
- Beland B, Fitzgerald M. Influence of peripheral inflammation on the postnatal maturation of primary sensory neuron phenotype in rats. J Pain. 2001 Feb;2(1):36–45. doi:https://doi.org/10.1054/jpai.2001.17697.
- Kupari J, Usoskin D, Lou D, et al. Single cell transcriptomics of primate sensory neurons identifies cell types associated with human chronic pain. bioRxiv. 2020. doi:https://doi.org/10.1101/2020.12.07.414193.
- Adelman PC, Baumbauer KM, Friedman R, Shah M, Wright M, Young E, Jankowski MP, Albers KM, Koerber HR. Single-cell q-PCR derived expression profiles of identified sensory neurons. Mol Pain Jan-Dec. 2019;15:1744806919884496. doi:https://doi.org/10.1177/1744806919884496.
- Jankowski MP, and Koerber HR. Frontiers in neuroscience neurotrophic factors and nociceptor Sensitization. In: Kruger L, and Light AR, editors. Translational pain research: from mouse to man. (2) Boca Raton, FL: CRC Press/Taylor & Francis Copyright © 2010 by Taylor and Francis Group, LLC. 1–34; 2010.
- Fariñas I, Wilkinson GA, Backus C, Reichardt LF, Patapoutian A. Characterization of neurotrophin and Trk receptor functions in developing sensory ganglia: direct NT-3 activation of TrkB neurons in vivo. Neuron. 1998 Aug;21(2):325–34. doi:https://doi.org/10.1016/s0896-6273(00)80542-5.
- Molliver DC, Wright DE, Leitner ML, Parsadanian AS, Doster K, Wen D, Yan Q, Snider WD. IB4-binding DRG neurons switch from NGF to GDNF dependence in early postnatal life. Neuron. 1997 Oct;19(4):849–61. doi:https://doi.org/10.1016/s0896-6273(00)80966-6.
- Dourson AJ, Ford ZK, Green KJ, McCrossan CE, Hofmann MC, Hudgins RC, Jankowski MP. Early life nociception is influenced by peripheral growth hormone signaling. J Neurosci. 2021 May 19;41(20):4410–27. doi:https://doi.org/10.1523/jneurosci.3081-20.2021.
- Chidambaran V, Zhang X, Geisler K, Stubbeman BL, Chen X, Weirauch MT, Meller J, Ji H. Enrichment of genomic pathways based on differential dna methylation associated with chronic postsurgical pain and anxiety in children: a prospective, pilot study. The Journal of Pain. 2019;20(7):771–85. doi:https://doi.org/10.1016/j.jpain.2018.12.008.
- Ford ZK, Dourson AJ, Liu X, Lu P, Green KJ, Hudgins RC, Jankowski MP. Systemic growth hormone deficiency causes mechanical and thermal hypersensitivity during early postnatal development. IBRO Reports. 2019 June 1;6:111–21. doi:https://doi.org/10.1016/j.ibror.2019.02.001.
- Dourson AJ, Ford ZK, Green KJ, McCrossan CE, Hofmann MC, Hudgins RC, Jankowski MP. Early life nociception is influenced by peripheral growth hormone signaling. J Neurosci. 2021 Apr 20;41(20):4410–27. doi:https://doi.org/10.1523/jneurosci.3081-20.2021.
- Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 2008 Jul 3;359(1):61–73. doi:https://doi.org/10.1056/NEJMra0708473.
- Quaife-Ryan GA, Sim CB, Ziemann M, Kaspi A, Rafehi H, Ramialison M, El-Osta A, Hudson JE, Porrello ER. Multicellular transcriptional analysis of mammalian heart regeneration. Circulation. 2017 Sep 19;136(12):1123–39. doi:https://doi.org/10.1161/circulationaha.117.028252.
- Fanucchi S, Domínguez-Andrés J, Joosten LAB, Netea MG, Mhlanga MM. The intersection of epigenetics and metabolism in trained immunity. Immunity. 2021 Jan 12;54(1):32–43. doi:https://doi.org/10.1016/j.immuni.2020.10.011.
- Netea MG, Joosten LA, Latz E, Mills KHG, Natoli G, Stunnenberg HG, O’Neill LAJ, Xavier RJ. Trained immunity: a program of innate immune memory in health and disease. Science. 2016 Apr 22;352(6284):aaf1098. doi:https://doi.org/10.1126/science.aaf1098.
- Cutfield WS, Hofman PL, Mitchell M, Morison IM. Could epigenetics play a role in the developmental origins of health and disease? Pediatr Res. 2007 May;61(5 Pt 2):68r–75r. doi:https://doi.org/10.1203/pdr.0b013e318045764c.
- Dudink J, Kerr JL, Paterson K, Counsell SJ. Connecting the developing preterm brain. Early Hum Dev. 2008 Dec;84(12):777–82. doi:https://doi.org/10.1016/j.earlhumdev.2008.09.004.
- Smith SB, Reenilä I, Männistö PT, Slade GD, Maixner W, Diatchenko L, Nackley AG. Epistasis between polymorphisms in COMT, ESR1, and GCH1 influences COMT enzyme activity and pain. Pain. 2014 Nov;155(11):2390–99. doi:https://doi.org/10.1016/j.pain.2014.09.009.
- Brewer CL, Baccei ML. The development of pain circuits and unique effects of neonatal injury. Journal of Neural Transmission (Vienna, Austria: 1996). 2019 Aug 9;127:467–79. doi:https://doi.org/10.1007/s00702-019-02059-z.
- Brewer CL, Li J, O’Conor K, Serafin EK, Baccei ML. Neonatal injury evokes persistent deficits in dynorphin inhibitory circuits within the adult mouse superficial dorsal horn. J Neurosci. 2020 May 13;40(20):3882–95. doi:https://doi.org/10.1523/jneurosci.0029-20.2020.
- Schwaller F, Beggs S, Walker SM. Targeting p38 mitogen-activated protein kinase to reduce the impact of neonatal microglial priming on incision-induced hyperalgesia in the adult rat. Anesthesiology. 2015 Jun;122(6):1377–90. doi:https://doi.org/10.1097/aln.0000000000000659.
- Moriarty O, Harrington L, Beggs S, Walker SM. Opioid analgesia and the somatosensory memory of neonatal surgical injury in the adult rat. Br J Anaesth. 2018 Jul;121(1):314–24. doi:https://doi.org/10.1016/j.bja.2017.11.111.
- Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001 Sep 13;413(6852):203–10. doi:https://doi.org/10.1038/35093019.
- Spencer SJ, Martin S, Mouihate A, Pittman QJ. Early-life immune challenge: defining a critical window for effects on adult responses to immune challenge. Neuropsychopharmacology. 2006 Sep;31(9):1910–18. doi:https://doi.org/10.1038/sj.npp.1301004.
- Zhong XS, Winston JH, Luo X, Kline KT, Nayeem SZ, Cong Y, Savidge TC, Dashwood RH, Powell DW, Li Q, et al. Neonatal colonic inflammation epigenetically aggravates epithelial inflammatory responses to injury in adult life. Cellular and Molecular Gastroenterology and Hepatology. 2018;6(1):65–78. doi:https://doi.org/10.1016/j.jcmgh.2018.02.014.
- Winterberg T, Vieten G, Meier T, Yu Y, Busse M, Hennig C, Hansen G, Jacobs R, Ure BM, Kuebler JF, et al. Distinct phenotypic features of neonatal murine macrophages. Eur J Immunol. 2015 Jan;45(1):214–24. doi:https://doi.org/10.1002/eji.201444468.
- Kumar SK, Bhat BV. Distinct mechanisms of the newborn innate immunity. Immunol Lett. 2016 May;173:42–54. doi:https://doi.org/10.1016/j.imlet.2016.03.009.
- Fragiadakis GK, Gaudilliere B, Ganio EA, Aghaeepour N, Tingle M, Nolan GP, Angst MS. Patient-specific immune states before surgery are strong correlates of surgical recovery. Anesthesiology. 2015 Dec;123(6):1241–55. doi:https://doi.org/10.1097/aln.0000000000000887.
- Shepherd AJ, Mickle AD, Golden JP, Mack MR, Halabi CM, de Kloet AD, Samineni VK, Kim BS, Krause EG, Gereau RW, et al. Macrophage angiotensin II type 2 receptor triggers neuropathic pain. Article. Proc Natl Acad Sci U S A. 2018;115(34):E8057–E8066. doi:https://doi.org/10.1073/pnas.1721815115.
- Chen S, Yang J, Wei Y, Wei X. Epigenetic regulation of macrophages: from homeostasis maintenance to host defense. Cell Mol Immunol. 2020 Jan;17(1):36–49. doi:https://doi.org/10.1038/s41423-019-0315-0.
- Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81(1):1–5. doi:https://doi.org/10.1189/jlb.0306164.
- Hong T, Donmez O, Miller D, et al. Epstein-Barr virus nuclear antigen 2 (EBNA2) extensively rewires the human chromatin landscape at autoimmune risk loci. bioRxiv. 2020. doi:https://doi.org/10.1101/2020.04.15.043612.
- Yoshida K, Maekawa T, Zhu Y, Renard-Guillet C, Chatton B, Inoue K, Uchiyama T, Ishibashi K-I, Yamada T, Ohno N, et al. The transcription factor ATF7 mediates lipopolysaccharide-induced epigenetic changes in macrophages involved in innate immunological memory. Nat Immunol. 2015 Oct 1;16(10):1034–43. doi:https://doi.org/10.1038/ni.3257.
- Laudanski K, Zawadka M, Polosak J, Modi J, DiMeglio M, Gutsche J, Szeto WY, Puzianowska-Kuznicka M. Acquired immunological imbalance after surgery with cardiopulmonary bypass due to epigenetic over-activation of PU.1/M-CSF. J Transl Med. 2018 [Accessed 2018 May];16(1):143. doi:https://doi.org/10.1186/s12967-018-1518-3.
- Bermick JR, Lambrecht NJ, denDekker AD, Kunkel SL, Lukacs NW, Hogaboam CM, Schaller MA. Neonatal monocytes exhibit a unique histone modification landscape. Clin Epigenetics. 2016;8(1):99. doi:https://doi.org/10.1186/s13148-016-0265-7.
- Reizel Y, Sabag O, Skversky Y, Spiro A, Steinberg B, Bernstein D, Wang A, Kieckhaefer J, Li C, Pikarsky E, et al. Postnatal DNA demethylation and its role in tissue maturation. Nat Commun. 2018 May 23;9(1):2040. doi:https://doi.org/10.1038/s41467-018-04456-6.
- Wallner S, Schroder C, Leitao E, Berulava T, Haak C, Beißer D, Rahmann S, Richter AS, Manke T, Bönisch U, et al. Epigenetic dynamics of monocyte-to-macrophage differentiation. Epigenetics Chromatin. 2016;9(1):33. doi:https://doi.org/10.1186/s13072-016-0079-z.
- Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007 Jun 21;447(7147):972–78. doi:https://doi.org/10.1038/nature05836.
- Fanucchi S, Dominguez-Andres J, Joosten LAB, Netea MG, Mhlanga MM. The intersection of epigenetics and metabolism in trained immunity. Immunity. 2021 Jan 12;54(1):32–43. doi:https://doi.org/10.1016/j.immuni.2020.10.011.
- Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009 Oct 16;139(2):267–84. doi:https://doi.org/10.1016/j.cell.2009.09.028.
- Pinho-Ribeiro FA, Baddal B, Haarsma R, O’Seaghdha M, Yang NJ, Blake KJ, Portley M, Verri WA, Dale JB, Wessels MR, et al. Blocking neuronal signaling to immune cells treats streptococcal invasive infection. Cell. 2018 May 17;173(5):1083–1097.e22. doi:https://doi.org/10.1016/j.cell.2018.04.006.
- Cohen JA, Edwards TN, Liu AW, Hirai T, Jones MR, Wu J, Li Y, Zhang S, Ho J, Davis BM, et al. Cutaneous TRPV1+ neurons trigger protective innate type 17 anticipatory immunity. Cell. 2019 Jul 19;178(4):919–932.e14. doi:https://doi.org/10.1016/j.cell.2019.06.022.
- Hoofwijk DM, van Reij RR, Rutten BP, Kenis G, Buhre WF, Joosten EA. Genetic polymorphisms and their association with the prevalence and severity of chronic postsurgical pain: a systematic review. Br J Anaesth. 2016 Dec;117(6):708–19. doi:https://doi.org/10.1093/bja/aew378.
- Chidambaran V, Gang Y, Pilipenko V, Ashton M, Ding L. Systematic review and meta-analysis of genetic risk of developing chronic postsurgical pain. J Pain. 2019 May 23. doi: https://doi.org/10.1016/j.jpain.2019.05.008.
- James SK. Chronic postsurgical pain: is there a possible genetic link? British Journal of Pain. 2017;11(4):178–85. doi:https://doi.org/10.1177/2049463717723222.
- Dimova V, Lotsch J, Huhne K, Winterpacht A, Heesen M, Parthum A, Weber PG, Carbon R, Griessinger N, Sittl R, et al. Association of genetic and psychological factors with persistent pain after cosmetic thoracic surgery. J Pain Res. 2015;8:829–44. doi:https://doi.org/10.2147/jpr.s90434.
- Thomazeau J, Rouquette A, Martinez V, Rabuel C, Prince N, Laplanche J-L, Nizard R, Bergmann J-F, Perrot S, Lloret-Linares C, et al. Predictive factors of chronic post-surgical pain at 6 months following knee replacement: influence of postoperative pain trajectory and genetics. Pain Physician. 2016 Jul; 19(5): E729–41.
- Kolesnikov Y, Gabovits B, Levin A, Veske A, Qin L, Dai F, Belfer I. Chronic pain after lower abdominal surgery: do catechol-o-methyl transferase/opioid receptor mu-1 polymorphisms contribute? Mol Pain. 2013 Apr 8;9:19. doi:https://doi.org/10.1186/1744-8069-9-19.
- Montes A, Roca G, Sabate S, Lao JI, Navarro A, Cantillo J, Canet J. Genetic and clinical factors associated with chronic postsurgical pain after hernia repair, hysterectomy, and thoracotomy: a two-year multicenter cohort study. Anesthesiology. 2015 May;122(5):1123–41. doi:https://doi.org/10.1097/aln.0000000000000611.
- Kalliomaki ML, Sandblom G, Hallberg M, Grönbladh A, Gunnarsson U, Gordh T, Ginya H, Nyberg F. Genetic susceptibility to postherniotomy pain. The influence of polymorphisms in the Mu opioid receptor, TNF-alpha, GRIK3, GCH1, BDNF and CACNA2D2 genes. Scand J Pain. 2016 Jul;12:1–6. doi:https://doi.org/10.1016/j.sjpain.2015.12.006.
- Hegarty D, Shorten G. Multivariate prognostic modeling of persistent pain following lumbar discectomy. Pain Physician. 2012 Sep-Oct;15(5):421–34. doi:https://doi.org/10.36076/ppj.2012/15/421.
- Hickey OT, Nugent NF, Burke SM, Hafeez P, Mudrakouski AL, Shorten GD. Persistent pain after mastectomy with reconstruction. J Clin Anesth. 2011 Sep;23(6):482–88. doi:https://doi.org/10.1016/j.jclinane.2011.01.009.
- Belfer I, Dai F, Kehlet H, Finelli P, Qin L, Bittner R, Aasvang EK. Association of functional variations in COMT and GCH1 genes with postherniotomy pain and related impairment. Pain. 2015 Feb;156(2):273–79. doi:https://doi.org/10.1097/01.j.pain.0000460307.48701.b0.
- Costigan M, Belfer I, Griffin RS, Dai F, Barrett LB, Coppola G, Wu T, Kiselycznyk C, Poddar M, Lu Y, et al. Multiple chronic pain states are associated with a common amino acid-changing allele in KCNS1. Brain. 2010 Sep;133(9):2519–27. doi:https://doi.org/10.1093/brain/awq195.
- Langford DJ, Paul SM, West CM, Dunn LB, Levine JD, Kober KM, Dodd MJ, Miaskowski C, Aouizerat BE. Variations in potassium channel genes are associated with distinct trajectories of persistent breast pain after breast cancer surgery. Pain. 2015 Mar;156(3):371–80. doi:https://doi.org/10.1097/01.j.pain.0000460319.87643.11.
- Wang L, Wei C, Xiao F, Chang X, Zhang Y. Influences of COMT rs4680 and OPRM1 rs1799971 polymorphisms on chronic postsurgical pain, acute pain, and analgesic consumption after elective cesarean delivery. Clin J Pain. 2019 Jan;35(1):31–36. doi:https://doi.org/10.1097/ajp.0000000000000654.
- Warner SC, van Meurs JB, Schiphof D, Bierma-Zeinstra SM, Hofman A, Uitterlinden AG, Richardson H, Jenkins W, Doherty M, Valdes AM, et al. Genome-wide association scan of neuropathic pain symptoms post total joint replacement highlights a variant in the protein-kinase C gene. Eur J Hum Genet. 2017 Apr;25(4):446–51. doi:https://doi.org/10.1038/ejhg.2016.196.
- van Reij RRI, Hoofwijk DMN, Rutten BPF, Weinhold L, Leber M, Joosten EAJ, Ramirez A, van Den Hoogen NJ. The association between genome-wide polymorphisms and chronic postoperative pain: a prospective observational study. Anaesthesia. 2020;75 Suppl 1(Suppl 1):e111–e120. doi:https://doi.org/10.1111/anae.14832.
- Stephens KE, Levine JD, Aouizerat BE, Paul SM, Abrams G, Conley YP, Miaskowski C. Associations between genetic and epigenetic variations in cytokine genes and mild persistent breast pain in women following breast cancer surgery. Cytokine. 2017 Nov;99:203–13. doi:https://doi.org/10.1016/j.cyto.2017.07.006.
- Blanc P, Génin E, Jesson B, Dubray C, and Dualé C. the E-gIg. Genetics and postsurgical neuropathic pain: an ancillary study of a multicentre survey. European Journal of Anaesthesiology | EJA. 2019;36(5):24.e1–24.e20.
- Packiasabapathy S, Horn N, Sadhasivam S. Genetics of perioperative pain management. Curr Opin Anaesthesiol. 2018;31(6):749–55. doi:https://doi.org/10.1097/ACO.0000000000000660.
- Sadhasivam S, Chidambaran V. Pharmacogenomics of opioids and perioperative pain management. Pharmacogenomics. 2012 Nov;13(15):1719–40. doi:https://doi.org/10.2217/pgs.12.152.
- Manworren RCB, Ruaño G, Young E, St Marie B, McGrath JM. Translating the human genome to manage pediatric postoperative pain. J Pediatr Surg Nurs Jan-Mar. 2015;4(1):28–39. doi:https://doi.org/10.1097/jps.0000000000000051.
- Rut M, Machoy-Mokrzyńska A, Ręcławowicz D, Słoniewski P, Kurzawski M, Droździk M, Safranow K, Morawska M, Białecka M. Influence of variation in the catechol-O-methyltransferase gene on the clinical outcome after lumbar spine surgery for one-level symptomatic disc disease: a report on 176 cases. Acta Neurochir (Wien). 2014;156(2):245–52. doi:https://doi.org/10.1007/s00701-013-1895-6.
- Dharaniprasad G, Samantaray A, Srikanth L, Hanumantha Rao M, Chandra A, Sarma P. Chronic persistent surgical pain is strongly associated with COMT alleles in patients undergoing cardiac surgery with median sternotomy. Gen Thorac Cardiovasc Surg. 2020 Oct;68(10):1101–12. doi:https://doi.org/10.1007/s11748-020-01321-6.
- Langford DJ, Paul SM, West CM, Dunn LB, Levine JD, Kober KM, Dodd MJ, Miaskowski C, and Aouizerat BE. Variations in potassium channel genes are associated with distinct trajectories of persistent breast pain after breast cancer surgery. Pain. 2015 Mar;156(3):371–380. doi: https://doi.org/10.1097/01.j.pain.0000460319.87643.11. PMID: 25599232
- Knisely MR, Conley YP, Kober KM, Smoot B, Paul SM, Levine JD, Miaskowski C. Associations between catecholaminergic and serotonergic genes and persistent breast pain phenotypes after breast cancer surgery. J Pain. 2018 Oct;19(10):1130–46. doi:https://doi.org/10.1016/j.jpain.2018.04.007.
- Nissenbaum J, Devor M, Seltzer Z, Gebauer M, Michaelis M, Tal M, Dorfman R, Abitbul-Yarkoni M, Lu Y, Elahipanah T, et al. Susceptibility to chronic pain following nerve injury is genetically affected by CACNG2. Genome Res. 2010 Sep;20(9):1180–90. doi:https://doi.org/10.1101/gr.104976.110.
- Yeo J, Sia AT, Sultana R, Sng BL, Tan EC. Analysis of SCN9A gene variants for acute and chronic postoperative pain and morphine consumption after total hysterectomy. Pain Med. 2020 Nov 1;21(11):2642–49. doi:https://doi.org/10.1093/pm/pnaa109.
- Dominguez C, Cui W. Inside out: decoding the transcriptome of effector and memory T cells. Immunol Cell Biol. 2013;91(6):389–90. doi:https://doi.org/10.1038/icb.2013.18.
- Ma G, Yang J, Zhao B, Huang C, Wang R. Correlation between CCL2, CALCA, and CX3CL1 gene polymorphisms and chronic pain after cesarean section in Chinese Han women: a case control study. Medicine. 2019;98(34):e16706–e16706. doi:https://doi.org/10.1097/MD.0000000000016706.
- Stephens K, Cooper BA, West C, Paul SM, Baggott CR, Merriman JD, Dhruva A, Kober KM, Langford DJ, Leutwyler H, et al. Associations between cytokine gene variations and severe persistent breast pain in women following breast cancer surgery. J Pain. 2014 Feb;15(2):169–80. doi:https://doi.org/10.1016/j.jpain.2013.09.015.
- Tegeder I, Costigan M, Griffin RS, Abele A, Belfer I, Schmidt H, Ehnert C, Nejim J, Marian C, Scholz J, et al. GTP cyclohydrolase and tetrahydrobiopterin regulate pain sensitivity and persistence. Nat Med. 2006 Nov;12(11):1269–77. doi:https://doi.org/10.1038/nm1490.
- Kim DH, Dai F, Belfer I, Banco RJ, Martha JF, Tighiouart H, Tromanhauser SG, Jenis LG, Hunter DJ, Schwartz CE, et al. Polymorphic variation of the guanosine triphosphate cyclohydrolase 1 gene predicts outcome in patients undergoing surgical treatment for lumbar degenerative disc disease. Spine (Phila Pa 1976). 2010;35(21):1909–14. doi:https://doi.org/10.1097/BRS.0b013e3181eea007.
- Weiskopf D, Bangs DJ, Sidney J, Kolla RV, De Silva AD, de Silva AM, Crotty S, Peters B, Sette A. Dengue virus infection elicits highly polarized CX3CR1 + cytotoxic CD4 + T cells associated with protective immunity. Proceedings of the National Academy of Sciences. 2015;112(31):E4256–E4263. doi:https://doi.org/10.1073/pnas.1505956112.
- Sia AT, Lim Y, Lim EP, Goh RC, Law H, Landau R, Teo -Y-Y, Tan E. A118G single nucleotide polymorphism of human μ-opioid receptor gene influences pain perception and patient-controlled intravenous morphine consumption after intrathecal morphine for postcesarean analgesia. Anesthesiology. 2008 Sep;109(3):520–26. doi:https://doi.org/10.1097/ALN.0b013e318182af21.
- Tian Y, Liu X, Jia M, Yu H, Lichtner P, Shi Y, Meng Z, Kou S, Ho IHT, Jia B, et al. Targeted genotyping identifies susceptibility locus in brain-derived neurotrophic factor gene for chronic postsurgical pain. Anesthesiology. 2018 Mar;128(3):587–97. doi:https://doi.org/10.1097/aln.0000000000001977.
- Yan S, Nie H, Bu G, Yuan W, Wang S. The effect of common variants in GDF5 gene on the susceptibility to chronic postsurgical pain. J Orthop Surg Res. 2021 Jul 1;16(1):420. doi:https://doi.org/10.1186/s13018-021-02549-5.
- Liu Y, Hou F, Wang X, Liu X. Recombinant expression and characterization of a serine protease inhibitor (Lvserpin7) from the Pacific white shrimp, Litopenaeus vannamei. Fish Shellfish Immunol. 2015 Feb;42(2):256–63. doi:https://doi.org/10.1016/j.fsi.2014.11.001.
- Antunes-Martins A, Perkins JR, Lees J, Hildebrandt T, Orengo C, Bennett DL. Systems biology approaches to finding novel pain mediators. Wiley Interdiscip Rev Syst Biol Med Jan-Feb. 2013;5(1):11–35. doi:https://doi.org/10.1002/wsbm.1192.
- Chidambaran MA V, Martin LJ, Jegga A. Systems biology-based approaches to summarize and identify novel genes and pathways associated with acute and chronic postsurgical pain. J Clin Anesth. 2020;62:109738. JCA_2019_1551_R2 (Accepted). doi:https://doi.org/10.1016/j.jclinane.2020.109738.
- Wood PB. Role of central dopamine in pain and analgesia. Review. Expert Rev Neurother. 2008;8(5):781–97. doi:https://doi.org/10.1586/14737175.8.5.781.
- van Reij RRI, Joosten EAJ, van Den Hoogen NJ. Dopaminergic neurotransmission and genetic variation in chronification of post-surgical pain. Br J Anaesth. 2019 Dec 1;123(6):853–64. doi:https://doi.org/10.1016/j.bja.2019.07.028.
- Lötsch J, Belfer I, Kirchhof A, Mishra BK, Max MB, Doehring A, Costigan M, Woolf CJ, Geisslinger G, Tegeder I, et al. Reliable screening for a pain-protective haplotype in the GTP cyclohydrolase 1 gene (GCH1) through the use of 3 or fewer single nucleotide polymorphisms. Article. Clin Chem. 2007;53(6):1010–15. doi:https://doi.org/10.1373/clinchem.2006.082883.
- Zorina-Lichtenwalter K, Meloto CB, Khoury S, Diatchenko L. Genetic predictors of human chronic pain conditions. Review. Neuroscience. 2016;338:36–62. doi:https://doi.org/10.1016/j.neuroscience.2016.04.041.
- Montes A, Roca G, Sabate S, Lao JI, Navarro A, Cantillo J, Canet J. Genetic and clinical factors associated with chronic postsurgical pain after hernia repair, hysterectomy, and thoracotomy: a two-year multicenter cohort study. article. Anesthesiology. 2015;122(5):1123–41. doi:https://doi.org/10.1097/ALN.0000000000000611.
- Megat S, Shiers S, Moy JK, Barragan-Iglesias P, Pradhan G, Seal RP, Dussor G, Price TJ. A critical role for dopamine d5 receptors in pain chronicity in male mice. J Neurosci. 2018 Jan 10;38(2):379–97. doi:https://doi.org/10.1523/jneurosci.2110-17.2017.
- Cobacho N, de La Calle JL, Paíno CL. Dopaminergic modulation of neuropathic pain: analgesia in rats by a D2-type receptor agonist. article. Brain Res Bull. 2014;106:62–71. doi:https://doi.org/10.1016/j.brainresbull.2014.06.003.
- Hoshino H, Obata H, Nakajima K, Mieda R, Saito S. The antihyperalgesic effects of intrathecal bupropion, a dopamine and noradrenaline reuptake inhibitor, in a rat model of neuropathic pain. Article. Anesth Analg. 2015;120(2):460–66. doi:https://doi.org/10.1213/ANE.0000000000000540.
- Jääskeläinen SK, Lindholm P, Valmunen T, Pesonen U, Taiminen T, Virtanen A, Lamusuo S, Forssell H, Hagelberg N, Hietala J, et al. Variation in the dopamine D2 receptor gene plays a key role in human pain and its modulation by transcranial magnetic stimulation. article. Pain. 2014;155(10):2180–87. doi:https://doi.org/10.1016/j.pain.2014.08.029.
- Andersen AM, Pietrzak RH, Kranzler HR, Ma L, Zhou H, Liu X, Kramer J, KupermanS, Edenberg, Nurnberger JI, et al. Polygenic scores for major depressive disorder and risk of alcohol dependencepolygenic risk score analysis for depression and alcohol dependencepolygenic risk score analysis for depression and alcohol dependence. JAMA Psychiatry. 2017;74(11):1153–60. doi:https://doi.org/10.1001/jamapsychiatry.2017.2269.
- Escott-Price V, Shoai M, Pither R, Williams J, Hardy J. Polygenic score prediction captures nearly all common genetic risk for Alzheimer’s disease. Neurobiol Aging. 2017 Jan 1;49:214.e7–214.e11. doi:https://doi.org/10.1016/j.neurobiolaging.2016.07.018.
- Muranen TA, Mavaddat N, Khan S, Fagerholm R, Pelttari L, Lee A, Aittomäki K, Blomqvist C, Easton DF, Nevanlinna H, et al. Polygenic risk score is associated with increased disease risk in 52 Finnish breast cancer families. Breast Cancer Res Treat. 2016;158(3):463–69. doi:https://doi.org/10.1007/s10549-016-3897-6.
- Richardson TG, Harrison S, Hemani G, Smith GD. An atlas of polygenic risk score associations to highlight putative causal relationships across the human phenome. Elife. 2019 Mar 5;8:ARTN e43657. doi:https://doi.org/10.7554/eLife.43657.
- Chidambaran V, Pilipenko V, Jegga AG, Geisler K, Martin LJ. systems biology guided gene enrichment approaches improve prediction of chronic post-surgical pain after spine fusion. original research. Front Genet. 2021 March 23;12(416). doi:https://doi.org/10.3389/fgene.2021.594250.
- van Reij RRI, Voncken JW, Joosten EAJ, van Den Hoogen NJ. Polygenic risk scores indicates genetic overlap between peripheral pain syndromes and chronic postsurgical pain. Neurogenetics. 2020 Jul;21(3):205–15. doi:https://doi.org/10.1007/s10048-020-00614-5.
- Gibson G. On the utilization of polygenic risk scores for therapeutic targeting. PLoS Genet. 2019;15(4):e1008060. doi:https://doi.org/10.1371/journal.pgen.1008060.
- Torkamani A, Wineinger NE, Topol EJ. The personal and clinical utility of polygenic risk scores. Nat Rev Genet. 2018 Sep 1;19(9):581–90. doi:https://doi.org/10.1038/s41576-018-0018-x.
- Denk F, McMahon SB, Tracey I. Pain vulnerability: a neurobiological perspective. Nat Neurosci. 2014 Feb;17(2):192–200. doi:https://doi.org/10.1038/nn.3628.
- Bie B, Brown DL, Naguib M. Synaptic plasticity and pain aversion. Eur J Pharmacol. 2011 Sep 30;667(1–3):26–31. doi:https://doi.org/10.1016/j.ejphar.2011.05.080.
- Chiechio S, Nicoletti F. Metabotropic glutamate receptors and the control of chronic pain. Curr Opin Pharmacol. 2012 Feb;12(1):28–34. doi:https://doi.org/10.1016/j.coph.2011.10.010.
- Denk F, McMahon SB. Chronic pain: emerging evidence for the involvement of epigenetics. Neuron. 2012 Feb 9;73(3):435–44. doi:https://doi.org/10.1016/j.neuron.2012.01.012.
- Doehring A, Geisslinger G, Lotsch J. Epigenetics in pain and analgesia: an imminent research field. European Journal of Pain (London, England). 2011 Jan;15(1):11–16. doi:https://doi.org/10.1016/j.ejpain.2010.06.004.
- Rahn EJ, Guzman-Karlsson MC, David Sweatt J. Cellular, molecular, and epigenetic mechanisms in non-associative conditioning: implications for pain and memory. Neurobiol Learn Mem. 2013 Oct;105:133–50. doi:https://doi.org/10.1016/j.nlm.2013.06.008.
- Mogil JS. Pain genetics: past, present and future. Trends Genet. 2012 Jun;28(6):258–66. doi:https://doi.org/10.1016/j.tig.2012.02.004.
- Buchheit T, Van de Ven T, Shaw A. Epigenetics and the transition from acute to chronic pain. Pain Med. 2012 Nov;13(11):1474–90. doi:https://doi.org/10.1111/j.1526-4637.2012.01488.x.
- Bai G, Ren K, Dubner R. Epigenetic regulation of persistent pain. Transl Res. 2015 Jan;165(1):177–99. doi:https://doi.org/10.1016/j.trsl.2014.05.012.
- Descalzi G, Ikegami D, Ushijima T, Nestler EJ, Zachariou V, Narita M. Epigenetic mechanisms of chronic pain. Trends Neurosci. 2015 Apr;38(4):237–46. doi:https://doi.org/10.1016/j.tins.2015.02.001.
- Machelska H, Celik MO. Recent advances in understanding neuropathic pain: glia, sex differences, and epigenetics. F1000Research. 2016;5:2743. doi:https://doi.org/10.12688/f1000research.9621.1.
- Garriga J, Laumet G, Chen S-R, Zhang Y, Madzo J, Issa JPJ, Pan H-L, Jelinek J. nerve injury-induced chronic pain is associated with persistent dna methylation reprogramming in dorsal root ganglion. J Neurosci. 2018 Jul 4;38(27):6090–101. doi:https://doi.org/10.1523/JNEUROSCI.2616-17.2018.
- Sun Y, Liang D, Sahbaie P, Clark JD. Effects of methyl donor diets on incisional pain in mice. PLoS One. 2013;8(10):e77881. doi:https://doi.org/10.1371/journal.pone.0077881.
- Sun Y, Sahbaie P, Liang DY, Li -W-W, Li X-Q, Shi X-Y, Clark JD. Epigenetic regulation of spinal CXCR2 signaling in incisional hypersensitivity in mice. Anesthesiology. 2013 Nov;119(5):1198–208. doi:https://doi.org/10.1097/ALN.0b013e31829ce340.
- López-Muñoz E, Mejía-Terrazas GE. Epigenetics and postsurgical pain: a scoping review. Pain Med. 2021 Jul 27. doi: https://doi.org/10.1093/pm/pnab234.
- Gonzalez-Jaramillo V, Portilla-Fernandez E, Glisic M, Voortman T, Ghanbari M, Bramer W, Chowdhury R, Nijsten T, Dehghan A, Franco OH, et al. Epigenetics and inflammatory markers: a systematic review of the current evidence. Int J Inflam. 2019;2019:6273680. doi:https://doi.org/10.1155/2019/6273680.
- Uhl GR, Sora I, Wang Z. The mu opiate receptor as a candidate gene for pain: polymorphisms, variations in expression, nociception, and opiate responses. Proc Natl Acad Sci U S A. 1999 Jul 6;96(14):7752–55. doi:https://doi.org/10.1073/pnas.96.14.7752.
- Hwang CK, Song KY, Kim CS, Choi HS, Guo X-H, Law P-Y, Wei L-N, Loh HH. Evidence of endogenous mu opioid receptor regulation by epigenetic control of the promoters. Mol Cell Biol. 2007 Jul;27(13):4720–36. doi:https://doi.org/10.1128/MCB.00073-07.
- Oertel BG, Doehring A, Roskam B, Kettner M, Hackmann N, Ferreirós N, Schmidt PH, Lötsch J. Genetic-epigenetic interaction modulates mu-opioid receptor regulation. Hum Mol Genet. 2012 Nov 1;21(21):4751–60. doi:https://doi.org/10.1093/hmg/dds314.
- Chorbov VM, Todorov AA, Lynskey MT, Cicero TJ. Elevated levels of DNA methylation at the OPRM1 promoter in blood and sperm from male opioid addicts. J Opioid Manag. 2011;7(4):258–64. Jul-Aug. doi:https://doi.org/10.5055/jom.2011.0067.
- Nielsen DA, Yuferov V, Hamon S, Jackson C, Ho A, Ott J, Kreek MJ. Increased OPRM1 DNA methylation in lymphocytes of methadone-maintained former heroin addicts. Neuropsychopharmacology. 2009 Mar;34(4):867–73. doi:https://doi.org/10.1038/npp.2008.108.
- Chidambaran V, Zhang X, Martin LJ, Ding L, Weirauch MT, Geisler K, Stubbeman BL, Sadhasivam S, Ji H. DNA methylation at the mu-1 opioid receptor gene (OPRM1) promoter predicts preoperative, acute, and chronic postsurgical pain after spine fusion. Pharmgenomics Pers Med. 2017;10:157–68. doi:https://doi.org/10.2147/PGPM.S132691.
- Kringel D, Kaunisto MA, Kalso E, Lotsch J. Machine-learned analysis of global and glial/opioid intersection-related DNA methylation in patients with persistent pain after breast cancer surgery. Clin Epigenetics. 2019 Nov 27;11(1):167. doi:https://doi.org/10.1186/s13148-019-0772-4.
- Chidambaran V, Zhang X, Geisler K, Stubbeman BL, Chen X, Weirauch MT, Meller J, Ji H. Enrichment of genomic pathways based on differential dna methylation associated with chronic postsurgical pain and anxiety in children: a prospective, pilot study. The Journal of Pain: Official Journal of the American Pain Society. 2019 Jan 9;20(7):771–85. doi:https://doi.org/10.1016/j.jpain.2018.12.008.
- Wu H, Huang Y, Tian X, Zhang Z, Zhang Y, Mao Y, Wang C, Yang S, Liu Y, Zhang W, et al. Preoperative anxiety-induced glucocorticoid signaling reduces GABAergic markers in spinal cord and promotes postoperative hyperalgesia by affecting neuronal PAS domain protein 4. Mol Pain Jan-Dec. 2019;15:1744806919850383–1744806919850383. doi:https://doi.org/10.1177/1744806919850383.
- Davies MN, Volta M, Pidsley R, Lunnon K, Dixit A, Lovestone S, Coarfa C, Harris RA, Milosavljevic A, Troakes C, et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012;13(6):R43. doi:https://doi.org/10.1186/gb-2012-13-6-r43.
- Frank MO. Circulating cell-free dna differentiates severity of inflammation. Biol Res Nurs. 2016 Oct;18(5):477–88. doi:https://doi.org/10.1177/1099800416642571.
- Chen L-Y, Qi J, Xu H-L, Lin X-Y, Sun Y-J, Ju S-Q. The value of serum cell-free dna levels in patients with schizophrenia. Frontiers in Psychiatry. 2021;12:637789. doi:https://doi.org/10.3389/fpsyt.2021.637789.
- Lehmann-Werman R, Neiman D, Zemmour H, Moss J, Magenheim J, Vaknin-Dembinsky A, Rubertsson S, Nellgård B, Blennow K, Zetterberg H, et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proceedings of the National Academy of Sciences. 2016;113(13):E1826–E1834. doi:https://doi.org/10.1073/pnas.1519286113.
- Levins KJ, Drago T, Roman E, Martin A, King R, Murphy P, Gallagher H, Barry D, O’Hanlon E, Roddy DW, et al. Magnetic resonance spectroscopy across chronic pain disorders: a systematic review protocol synthesising anatomical and metabolite findings in chronic pain patients. Syst Rev. 2019 Dec 27;8(1):338. doi:https://doi.org/10.1186/s13643-019-1256-5.
- Buchheit T, Van de Ven T, Shaw A. Epigenetics and the Transition from Acute to Chronic Pain. Pain Med. 2012;13(11):1474–90. doi:https://doi.org/10.1111/j.1526-4637.2012.01488.x.
- Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet. 2011 Jul 12;12(8):529–41. doi:https://doi.org/10.1038/nrg3000.
- Boks MP, Derks EM, Weisenberger DJ, Strengman E, Janson E, Sommer IE, Kahn RS, Ophoff RA. The relationship of DNA methylation with age, gender and genotype in twins and healthy controls. PLoS One. 2009 Aug 26;4(8):e6767. doi:https://doi.org/10.1371/journal.pone.0006767.
- Niculescu AB, Le-Niculescu H, Levey DF, Roseberry K, Soe KC, Rogers J, Khan F, Jones T, Judd S, McCormick MA, et al. Towards precision medicine for pain: diagnostic biomarkers and repurposed drugs. Mol Psychiatry. 2019 Apr 1;24(4):501–22. doi:https://doi.org/10.1038/s41380-018-0345-5.
- Sisignano M, Lötsch J, Parnham MJ, Geisslinger G. Potential biomarkers for persistent and neuropathic pain therapy. Pharmacol Ther. 2019 Jul;199:16–29. doi:https://doi.org/10.1016/j.pharmthera.2019.02.004.
- Chidambaran V, Zhang X, Pilipenko V, Chen X, Wronowski B, Geisler K, Martin LJ, Barski A, Weirauch MT, Ji H, et al. Methylation quantitative trait locus analysis of chronic postsurgical pain uncovers epigenetic mediators of genetic risk. Epigenomics. 2021 Apr;13(8):613–30. doi:https://doi.org/10.2217/epi-2020-0424.
- Lenzenweger MF. Thinking clearly about the endophenotype–intermediate phenotype–biomarker distinctions in developmental psychopathology research. Dev Psychopathol. 2013;25(4pt2):1347–57. doi:https://doi.org/10.1017/S0954579413000655.
- Lebe M, Hasenbring MI, Schmieder K, Jetschke K, Harders A, Epplen JT, Hoffjan S, Kötting J. Association of serotonin-1A and −2A receptor promoter polymorphisms with depressive symptoms, functional recovery, and pain in patients 6 months after lumbar disc surgery. Pain. 2013 Mar;154(3):377–84. doi:https://doi.org/10.1016/j.pain.2012.11.017.
- George SZ, Wu SS, Wallace MR, Moser MW, Wright TW, Farmer KW, Greenfield WH, Dai Y, Li H, Fillingim RB, et al. Biopsychosocial influence on shoulder pain: influence of genetic and psychological combinations on twelve-month postoperative pain and disability outcomes. Arthritis Care Res (Hoboken). 2016 Nov;68(11):1671–80. doi:https://doi.org/10.1002/acr.22876.
- Bonin RP, De Koninck Y. Restoring ionotropic inhibition as an analgesic strategy. Neurosci Lett. 2013 Dec 17;557(Pt A):43–51. doi:https://doi.org/10.1016/j.neulet.2013.09.047.
- Ossipov MH, Morimura K, Porreca F. Descending pain modulation and chronification of pain. Curr Opin Support Palliat Care. 2014 Jun;8(2):143–51. doi:https://doi.org/10.1097/SPC.0000000000000055.
- Peirs C, Seal RP. Neural circuits for pain: recent advances and current views. Science. 2016 Nov 4;354(6312):578–84. doi:https://doi.org/10.1126/science.aaf8933.
- Kuner R, Flor H. Structural plasticity and reorganisation in chronic pain. Nat Rev Neurosci. 2016 Dec 15;18(1):20–30. doi:https://doi.org/10.1038/nrn.2016.162.
- Wulff H, Christophersen P, Colussi P, Chandy KG, Yarov-Yarovoy V. Antibodies and venom peptides: new modalities for ion channels. Nat Rev Drug Discov. 2019 May;18(5):339–57. doi:https://doi.org/10.1038/s41573-019-0013-8.
- Moreno AM, Aleman F, Catroli GF, Hunt M, Hu M, Dailamy A, Pla A, Woller SA, Palmer N, Parekh U, et al. 2021 Mar 10. Long-lasting analgesia via targeted in situ repression of Na V 1.7 in mice. Sci Transl Med; 13(584).doi: https://doi.org/10.1126/scitranslmed.aay9056
- Barragan-Iglesias P, Lou TF, Bhat VD, Megat S, Burton MD, Price TJ, Campbell ZT. Inhibition of Poly(A)-binding protein with a synthetic RNA mimic reduces pain sensitization in mice. Nat Commun. 2018 Jan 2;9(1):10. doi:https://doi.org/10.1038/s41467-017-02449-5.
- Mamet J, Klukinov M, Yaksh TL, Malkmus SA, Williams S, Harris S, Manning DC, Taylor BK, Donahue RR, Porreca F, et al. Single intrathecal administration of the transcription factor decoy AYX1 prevents acute and chronic pain after incisional, inflammatory, or neuropathic injury. Pain. 2014 Feb;155(2):322–33. doi:https://doi.org/10.1016/j.pain.2013.10.015.
- Sun Y, Sahbaie P, Liang D, Li W, Shi X, Kingery P, Clark JD. DNA methylation modulates nociceptive sensitization after incision. PLoS One. 2015;10(11):e0142046. doi:https://doi.org/10.1371/journal.pone.0142046.
- Alvarado S, Tajerian M, Suderman M, Machnes Z, Pierfelice S, Millecamps M, Stone LS, Szyf M. An epigenetic hypothesis for the genomic memory of pain. hypothesis and theory. Front Cell Neurosci. 2015 March 24;9(88). doi:https://doi.org/10.3389/fncel.2015.00088.
- Szyf M. Epigenetics, DNA methylation, and chromatin modifying drugs. Annu Rev Pharmacol Toxicol. 2009;49:243–63. doi:https://doi.org/10.1146/annurev-pharmtox-061008-103102.
- Maeder ML, Angstman JF, Richardson ME, Linder SJ, Cascio VM, Tsai SQ, Ho QH, Sander JD, Reyon D, Bernstein BE, et al. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat Biotechnol. 2013 Dec;31(12):1137–42. doi:https://doi.org/10.1038/nbt.2726.
- Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014 Jun 5;157(6):1262–78. doi:https://doi.org/10.1016/j.cell.2014.05.010.
- Sumizono M, Sakakima H, Otsuka S, Terashi T, Nakanishi K, Ueda K, Takada S, Kikuchi K. The effect of exercise frequency on neuropathic pain and pain-related cellular reactions in the spinal cord and midbrain in a rat sciatic nerve injury model. J Pain Res. 2018;11:281–91. doi:https://doi.org/10.2147/JPR.S156326.
- Kami K, Taguchi S, Tajima F, Senba E. histone acetylation in microglia contributes to exercise-induced hypoalgesia in neuropathic pain model mice. J Pain. 2016 May;17(5):588–99. doi:https://doi.org/10.1016/j.jpain.2016.01.471.
- Kaye AD, Koress CM, Novitch MB, Jung JW, Urits I, Viswanath O, Renschler JS, Alpaugh ES, Cornett EM. Pharmacogenomics, concepts for the future of perioperative medicine and pain management: a review. Best Pract Res Clin Anaesthesiol. 2020 Sep 1;34(3):651–62. doi:https://doi.org/10.1016/j.bpa.2020.07.004.
- Landau R, Kern C, Columb MO, Smiley RM, Blouin J-L. Genetic variability of the μ-opioid receptor influences intrathecal fentanyl analgesia requirements in laboring women. PAIN. 2008 Sep 30;139(1):5–14. doi:https://doi.org/10.1016/j.pain.2008.02.023.
- Choudhary J, Grant SG. Proteomics in postgenomic neuroscience: the end of the beginning. Nat Neurosci. 2004 May;7(5):440–45. doi:https://doi.org/10.1038/nn1240.
- Gomez-Varela D, Barry AM, Schmidt M. Proteome-based systems biology in chronic pain. J Proteomics. 2019 Jan 6;190:1–11. doi:https://doi.org/10.1016/j.jprot.2018.04.004.
- Olausson P, Gerdle B, Ghafouri N, Larsson B, Ghafouri B. Identification of proteins from interstitium of trapezius muscle in women with chronic myalgia using microdialysis in combination with proteomics. PLoS One. 2012;7(12):e52560. doi:https://doi.org/10.1371/journal.pone.0052560.
- Muntel J, Xuan Y, Berger ST, Reiter L, Bachur R, Kentsis A, Steen H. Advancing urinary protein biomarker discovery by data-independent acquisition on a quadrupole-orbitrap mass spectrometer. J Proteome Res. 2015 Nov 6;14(11):4752–62. doi:https://doi.org/10.1021/acs.jproteome.5b00826.
- Ghafouri B, Carlsson A, Holmberg S, Thelin A, Tagesson C. Biomarkers of systemic inflammation in farmers with musculoskeletal disorders; a plasma proteomic study. BMC Musculoskelet Disord. 2016 May 10;17:206. doi:https://doi.org/10.1186/s12891-016-1059-y.
- Kronfol MM, Dozmorov MG, Huang R, Slattum PW, McClay JL. The role of epigenomics in personalized medicine. Expert Review of Precision Medicine and Drug Development. 2017;2(1):33–45. doi:https://doi.org/10.1080/23808993.2017.1284557.
- Iskandar BJ, Rizk E, Meier B, Hariharan N, Bottiglieri T, Finnell RH, Jarrard DF, Banerjee RV, Skene JHP, Nelson A, et al. Folate regulation of axonal regeneration in the rodent central nervous system through DNA methylation. J Clin Invest. 2010 May;120(5):1603–16. doi:https://doi.org/10.1172/jci40000.
- Kanherkar RR, Stair SE, Bhatia-Dey N, Mills PJ, Chopra D, Csoka AB. Epigenetic mechanisms of integrative medicine. Evid Based Complement Alternat Med. 2017;2017:19. 4365429. doi:https://doi.org/10.1155/2017/4365429.
- García-Campayo J, Puebla-Guedea M, Labarga A, Urdánoz A, Roldán M, Pulido L, de Morentin XM, perdones-montero á, montero-marín j, mendioroz m, et al. epigenetic response to mindfulness in peripheral blood leukocytes involves genes linked to common human diseases. journal article. Mindfulness. 2018 August 01;9(4):1146–59. doi:https://doi.org/10.1007/s12671-017-0851-6.
- Stahl SM. Psychotherapy as an epigenetic ‘drug’: psychiatric therapeutics target symptoms linked to malfunctioning brain circuits with psychotherapy as well as with drugs. J Clin Pharm Ther. 2012 Jun;37(3):249–53. doi:https://doi.org/10.1111/j.1365-2710.2011.01301.x.