
I would like to start this talk by reminding you who Dr. James Burns Amberson was and why this lecture is named after him. Dr. Amberson went to medical school just down the road at Johns Hopkins Medical School but in his final year he was diagnosed with tuberculosis (TB) and spent the next several years in the Loomis sanatorium in New York State. The illness kindled an interest in lung disease and he went on to a career in Pulmonary Medicine, becoming the president of the American Thoracic Society (ATS) in 1940 and winning the Trudeau Medal in 1952. He was head of the Chest Service at Columbia University between 1938 and 1955.
I thought I would begin this talk with acknowledgments since these are often given short shrift at the end of a talk that goes on too long. I have had enormous good fortune in my career to have outstanding mentors, colleagues, trainees and collaborators.
First and foremost, I thank my primary mentor and friend Jim Hogg with whom I have worked throughout my career. Jim is a continuous source of ideas, inspiration and fun. The only trouble is he is hard to keep up with!
I had other mentors while at McGill including Peter Macklem and Nick Anthonisen, as well as David Bates, Margot Becklake, Ludwig Engel, Milic Emili, Paul Despas and Whitey Thurlbeck.
At McGill and UBC (University of British Columbia), I have had a wonderful group of colleagues and the greatest joy in my career has been the trainees. We have been lucky enough to have a steady stream of wonderful students and post docs from around the world in our laboratory. It is their curiosity, questions and drive that have fueled my research and they have become friends and collaborators. I would like to thank all of my collaborators; modern science is a collective effort and the opportunity to work with such a diverse group of passionate experts has been very rewarding.
Finally, I want to thank my most important collaborator…in science and life…Lisa Baile… we trained and worked together, but Lisa has made sure I experience life beyond Respiratory Medicine.
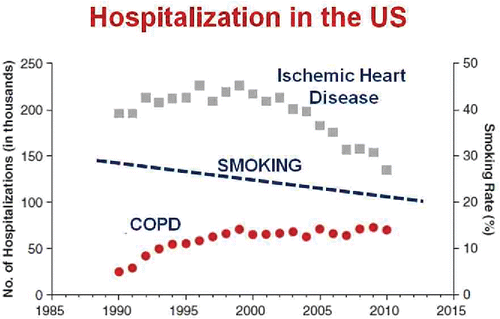
Now to tell you what we have been working on recently. I will start with the good news and the bad news. First the bad: according to recent evidence, we are all going to die!!! The good news, however, is that we are much less likely to die of cardiovascular disease now than we were 30 or 40 years ago. Wilmot et alCitation1 showed that there has been a dramatic decline in mortality from cardiovascular disease in the United States since 1979; a halving of mortality for both men and women. This decline in mortality is mirrored by a decline in hospitalization for ischemic heart disease in the United States (), which is a trend that is not shared for COPD.Citation2 This is despite a continued decrease in the prevalence of smoking. In Canada, COPD exacerbations are now the most common reason for hospitalization.Citation3
Figure 1. Number of hospitalizations for ischemic heart disease and COPD in the United States between 1985 and 2010 (left vertical axis) and the percentage of the population who smoked cigarettes (right vertical axis) over the same time period. There has been a progressive reduction in admissions for heart disease that is not mirrored for COPD despite a continuous reduction in the prevalence of smoking. Adapted from Reference 2 and reprinted with permission of the American Thoracic Society. Copyright (c) 2017 American Thoracic Society. Khakban A, Sin DD, FitzGerald JM, McManus BM, Ng R, Hollander Z, Sadatsafavi M. The Projected Epidemic of Chronic Obstructive Pulmonary Disease Hospitalizations over the Next 15 Years. A Population-based Perspective. Am J Respir Crit Care Med. 2017 Feb 1;195(3):287–291. The American Journal of Respiratory and Critical Care Medicine is an official journal of the American Thoracic Society.
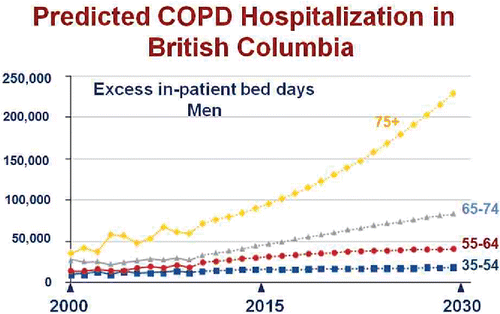
Colleagues at UBC have modeled this trend to predict the future impact of COPD on hospitalizations in the province of British Columbia.Citation2 Their model () predicts that hospitalizations will continue to increase, especially for the older age group. This dramatic result is driven, in part, by an increase in the fraction of the population over 75 years of age and by population growth; however, an additional reason is our failure to develop disease modifying therapy for COPD beyond smoking cessation.
Figure 2. Actual (solid line) and projected (dotted line) in-patient hospitalization days for exacerbations of COPD in the Province of British Columbia between 2000 and 2030 stratified by age group. Adapted from Reference 2 and reprinted with permission of the American Thoracic Society. Copyright (c) 2017 American Thoracic Society. Khakban A, Sin DD, FitzGerald JM, McManus BM, Ng R, Hollander Z, Sadatsafavi M. The Projected Epidemic of Chronic Obstructive Pulmonary Disease Hospitalizations over the Next 15 Years. A Population-based Perspective. Am J Respir Crit Care Med. 2017 Feb 1;195(3):287–291. The American Journal of Respiratory and Critical Care Medicine is an official journal of the American Thoracic Society.
The dramatic decline in cardiovascular disease mortality and hospitalization is due to a number of factors including widespread public health measures to change behavior and the treatment of acute cardiovascular events; however, a major factor is evidence-based drug therapy for primary and secondary prevention developed as a consequence of knowledge about the basic molecular pathophysiology of Atherosclerosis; therapy aimed at lowering blood pressure, hyperglycemia and lipids.Citation4 Cardiovascular scientists developed this disease-modifying therapy because of knowledge gained from careful experimental studies of animal models and the human disease, as well as the occurrence of single gene disorders that dramatically increase the risk of atherosclerosis. Our therapies for COPD are aimed at symptom relief. We need to understand the basic molecular pathophysiology of COPD to design novel therapies. To date, the careful pathological studies of COPD led by my colleague Jim HoggCitation5,6 have taught us a lot about the nature of the lesions in the peripheral airways that initiate COPD, but neither of these studies nor extrapolation from single gene disorders, such as alpha one antitrypsin deficiency, have led to treatment interventions that can alter the natural history of the disease in the vast majority of those with COPD.
Do genetics and genomics offer a pathway to this aim? Francis Collins thought so. With the completion of the human genome project he promised that it would be, “a transformative textbook of medicine, with insights that will give health care providers immense new powers to treat, prevent and cure disease” (https://www.genome.gov/11511417/). Has it delivered?
One thing that the Human Genome Project has enabled is the ability to conduct Genome Wide Association Studies (GWAS) (). To do a GWAS, one needs to gather a large number of individuals with the disease of interest and an equally large group of individuals matched for demographic characteristics other than the presence of the disease who are the controls. One obtains DNA from all individuals and assays one million or more genotypes at polymorphic sites across the genome on each individual. The vast majority of these are single nucleotide polymorphisms (SNPs). One then compares the frequency of individual genotypes at these one million or more loci in the cases and controls and calculates the odds ratio and p value of being a case versus a control based on those one million genotypes. In the example shown in , cases have a significant enrichment for the A allele compared to the controls.
Figure 3. Genome Wide Association Studies (GWAS) require a large number of cases matched for demographics with a large number of controls. Each individual is genotyped at one million or more polymorphic sites in their genome and the distribution of genotypes at each locus is compared in cases and controls. Finding a genotype whose frequency is different between cases and controls with a p value < 5 × 10−8 indicates a susceptibility locus for the disease (ie, a genetic variant that increases risk).

The results of a GWAS can be displayed using a Manhattan plot (). This shows all one million plus polymorphisms as single colored spots ordered by chromosomal location versus the negative log10 of the p value for association with a specific phenotype. Given the enormous number of tests performed, the convention is that a significant association occurs when the p value is equal to or less than 5 × 10−8 as shown by the dotted red line in this graph.
Figure 4. The results of GWAS are depicted as a Manhattan plot, which is named as such because they resemble the Manhattan skyline. Each polymorphic DNA site (mainly single nucleotide polymorphisms [SNPs]) is shown with a colored dot ordered by location on the chromosomes versus the negative log 10 p value that is associated with the trait or disease of interest. The red dotted line is at 5 × 10−8 and is the conventional threshold for significance given the large number of tests that are performed.
![Figure 4. The results of GWAS are depicted as a Manhattan plot, which is named as such because they resemble the Manhattan skyline. Each polymorphic DNA site (mainly single nucleotide polymorphisms [SNPs]) is shown with a colored dot ordered by location on the chromosomes versus the negative log 10 p value that is associated with the trait or disease of interest. The red dotted line is at 5 × 10−8 and is the conventional threshold for significance given the large number of tests that are performed.](/cms/asset/afe67f47-5f9e-409f-b3b9-302402268913/ucts_a_1361203_f0004_c.gif)
Genome-wide association studies have proliferated exponentially. The NIH maintains an online catalog of GWAS results and as of 12 June 2017, the GWAS Catalog contained 2974 publications and 36,618 unique SNP-trait associations (https://www.ebi.ac.uk/gwas/). The list of traits and diseases, where one or more SNP have achieved the genome-wide threshold for significant association, includes hair color and freckles but also respiratory diseases and phenotypes including Asthma,Citation7 COPD,Citation8,9 Lung function,Citation10 CF disease severityCitation11 and IPF.Citation12 As a result, GWAS WORKS!
We were fortunate to be involved in one of the largest GWAS studies of asthma to date.Citation7 The Gabriel study recruited over 10 000 cases with physician diagnosed asthma and 16 000 controls from a number of countries. Eight locations achieved genome-wide significance and the names of the genes closest to the polymorphisms were: IL18R1, ST-2, TSLP, IL13, IL33, HLA-DQ, SMAD3, ORMDL3/GSDMB/GSDMA and IL2RB. Interestingly, when a GWAS was done using total IgE as the phenotype in the same individuals, the only locus that overlapped was the IL13 locus suggesting that there are separate genetic determinants of asthma and atopy.
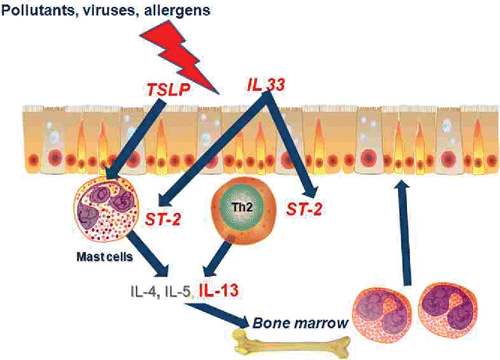
Further examination of the GWAS hits revealed some familiar candidates such as IL13, TSLP, IL33 and ST2. These had been previously postulated to be involved in the pathogenesis of asthma and, thus, it was gratifying that individuals who harbored genetic variants for these genes had altered susceptibility. A closer examination of the GWAS hits suggested a new theory for the origin of asthma. Three of the genes, TSLP, IL33 and GSDMA, are expressed in airway epithelium and ST2 is the receptor for IL33 expressed on inflammatory/immune cells. Interpretation of these results suggested a new paradigm for the molecular pathogenesis of asthma as shown in , which depicts how these susceptibility genes could interact to enhance allergic airway inflammation. External environmental agents including allergens, viruses and pollutants can damage the airway epithelium inducing the release of TSLP and IL33. TSLP can act on inflammatory/immune cells, including mast cells to induce release of Th2 cytokines and IL33 can similarly act on mast cells and Th2 lymphocytes via its receptor ST2 to release the same Th2 cytokines. The Th2 cytokines, including IL13, can act directly on lung tissue cells including the epithelium and smooth muscle and also on the bone marrow to release inflammatory cells and direct them to the airway epithelium. Thus, the action and interaction of 4 of the susceptibility genes for asthma can initiate an epithelial-cell based Th2-type airway inflammation without the involvement of IgE. This paradigm implicates the airway epithelial cell as the conductor of the asthma “orchestra” rather than immune/inflammatory cells.
Figure 5. A schema for epithelial-centric origin of asthma. Environmental factors including viruses, pollution and allergens cause release of TSLP and IL33 from airway epithelium. TSLP and IL33 (via ST-2) stimulate the release of Th2 cytokines (including IL13) that have a direct effect on structural airway cells and stimulate the release of inflammatory cells from the bone marrow. The genes for which genetic variants alter susceptibility for asthma are indicated in red text.
Have these results proved useful clinically? Related in part to the GWAS results, two novel therapies for asthma that target pathways influenced by susceptibility genes have been developed over the last few years. Gauvreau and colleaguesCitation13 (13) tested a monoclonal antibody designed to block the action of TSLP on the response to acute allergen inhalation in allergic asthmatics. They found that after 6 weeks of therapy there was a significant attenuation in both the acute and delayed airway response to inhaled allergens; the beneficial effect persisted till 12 weeks, which was the last measurement time. This new therapy is presently in Phase 2 clinical trials.
Sally Wenzel and colleaguesCitation14 tested the effect of a monoclonal antibody Dupilumab which blocks the IL4 and IL13 receptor, on exacerbation rate during an 8-week period of slow withdrawal of therapy in asthmatics. Blocking the action of these genes, which have been implicated in asthma GWAS, dramatically reduced asthma exacerbations. Only 6% of subjects on Dupilumab had exacerbations over the 8-week period compared to 44% on placebo (an 87% reduction; odds ratio, 0.08; 95% confidence interval, 0.02 to 0.28; P < 0.001). This therapy has been recently approved by the FDA for Atopic Dermatitis in which it is equally effective and approval for asthma is likely to occur soon.
A key feature of these results is that the beneficial effects of TSLP and IL13 blockade were experienced by most of the subjects who received the treatment…not just the carriers of the susceptibility genotypes. This shows that the genetic analyses can identify pathophysiological pathways that are important not only in carriers, but in most who have a disease. In a way, this is the antithesis of personalized medicine…a therapy identified by way of susceptibility in a minority that is beneficial for the majority.
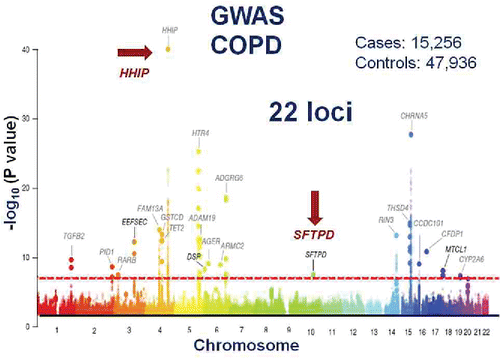
How about the GWAS results for lung function and COPD? A number of GWAS have been performed for both.Citation8,9,10 shows the results for Forced expiratory volume/forced vital capacity (FEV1/FVC) from one large study of lung function in a general population of smokers and non-smokers.Citation10 A total of 26 loci reached the genome wide threshold. The vast majority where never considered as putative candidate genes for COPD. There are no obvious biological patterns, although two of the genes implicated, Hedge Hog Interacting Protein (HHIP) and Patched 1 (PTCH1), are both in the hedgehog signaling pathway; HHIP is a negative regulator of the pathway and PCTH1 is the receptor that activates the pathway. I will return to these later in the talk. A recent large GWAS for COPD published by Hobbs and associatesCitation8 revealed a similar number of “hits”; 22 independent loci, 13 of which overlapped with the lung function-related susceptibility genes (). Again, no obvious biological pathways were evident, but HHIP was the most significant hit, and a lung protein Surfactant Protein D achieved the genome wide threshold; I will return to it as well.
Figure 6. A Manhattan plot for a large GWAS of lung function. A total of 26 loci achieved genome-wide significance for association with FEV1% predicted and/or FEV1/FVC%. Data for FEV1/FVC% are shown. Most genes were not a priori candidates for low lung function. Two genes, HHIP and PTCH1 are indicated with arrows and are in the hedgehog signaling pathway. See text for more discussion of these genes. Reprinted by permission from Macmillan Publishers Ltd: Soler Artigas M, Loth DW, Wain LV, et al. Genome-wide association and large-scale follow up identifies 16 new loci influencing lung function. Nat Genet. 2011 Sep 25;43(11):1082–1090.
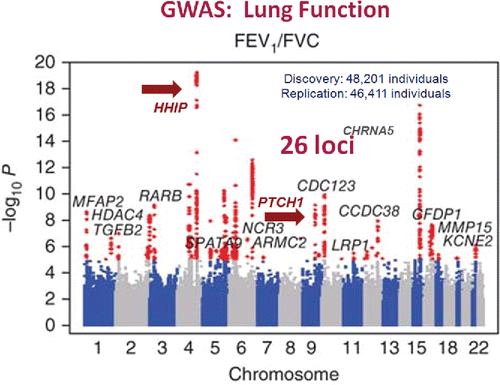
Figure 7. A Manhattan plot for a large GWAS of COPD. A total of 22 loci achieved genome wide significance for association with COPD in this case control design. HHIP, which was also identified as a susceptibility locus for low lung function, is indicated by the red arrow and was the most significant “hit.” Reprinted by permission from Macmillan Publishers Ltd: Hobbs BD, de Jong K, Lamontagne M, et al. Genetic loci associated with chronic obstructive pulmonary disease overlap with loci for lung function and pulmonary fibrosis. Nat Genet. 2017 Mar;49(3):426–432.
The challenge with such results is how to prioritize the susceptibility genes for future study and tease out how they influence risk for low lung function and COPD. Despite the lack of immediately obvious biologically plausible candidates (based on our limited knowledge), we should be encouraged by the results from asthma. These GWAS hits point to pathways important in COPD pathogenesis and targeting these pathways may have significant benefit.
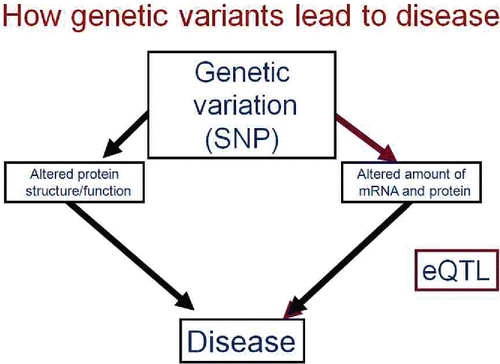
As shown in , gene variants can influence lung function by altering the structure, function or amount of its protein product. If the sequence variation is in the coding region of the gene, it can change amino acid sequence and produce an altered protein. Alternatively, it can alter a regulatory region such that it changes the level of expression of the gene product. Such loci are termed expression quantitative trait loci (eQTL), a cumbersome term, but simply a polymorphic DNA site that influences the expression of a gene. Such SNPs occur, not in the coding region of the gene, but in introns or inter-genic regions where they can alter a promoter, repressor, enhancer or methylation site to change the baseline or stimulated level of expression of a nearby gene or genes.
Figure 8. Genetic variation can affect phenotype by altering the amino acid sequence and, thus, the structure or function of the coded protein, or can affect sequences that influence the quantity of mRNA and thus protein produced. The latter are termed expression quantitative trait loci or eQTLs.
To date, most susceptibility loci for common diseases discovered by GWAS have been eQTLs;Citation15,16 as a result, we hypothesized that SNPs that influence lung function and risk for lung disease do so by modulating the level of gene expression in the lung. To test this, we first had to study the genetic control of gene expression in the lung.
This was achieved through a collaborative effort between investigators at UBC, Laval University, the University of Groningen and Merck.Citation17 We assayed 2.5 million SNPs in more than eleven hundred individuals and measured genome-wide gene expression in a sample of previously frozen lung tissue from the same 1100 individuals.
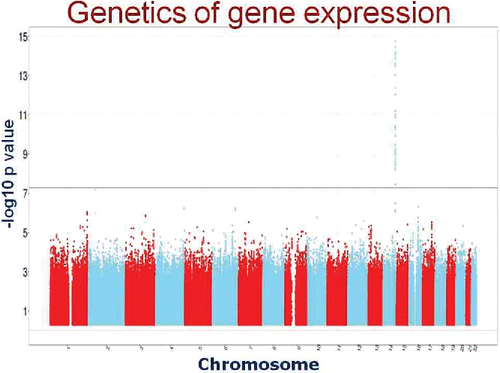
We then associated the SNPs with gene expression. Basically, we performed 25 000 GWAS with the phenotype being the level of individual gene expression. The results of one such analysis that showed an eQTL is shown as a Manahattan plot in . As can be seen, there is only one region of the genome that significantly influences the expression of this gene on Chromosome 14, and the SNPs that influence its expression are located close to the gene…a so-called cis or local eQTL.
Figure 9. Typical Manhattan plot for a cis, or local, eQTL. SNPs on chromosome 14 are highly significantly associated with the level of gene expression of a nearby gene.
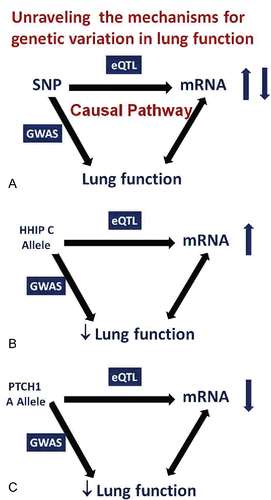
We found that almost half of the human genes expressed in the lung had SNPs that control their expression level using a 10% FDR cut off (P ≤ 1 × 10−6). The next step in unraveling how the genetic variants influence lung function was performed by Maen Obeidat in our lab.Citation18 He tested whether the SNPs that were associated with lung function also influenced gene expression in the lung (increased or decreased mRNA) and finally he tested whether there was a relationship between the level of expression and lung function (). For example, as shown earlier, SNPs near the HHIP gene were associated with low lung function and COPD. The same SNPs cause an increase in the lung tissue expression of HHIP, and higher HHIP mRNA in lung tissue is associated with lower lung function (). This suggests that increased HHIP is detrimental for lung function. The other gene in the hedgehog pathway that was a GWAS hit for low lung function was PTCH1, the Hedgehog pathway receptor. The SNPs associated with low lung function also influenced the level of gene expression but, in this instance, were associated with lower mRNA (). Similarly, individuals with low tissue mRNA for PTCH1 had lower lung function.
Figure 10. A. Unraveling the causal pathway by which gene variation influences phenotype. If a SNP is a risk factor for a trait AND the same SNP alters the level of expression of a gene, this suggests that the increased risk is caused by altered gene expression. This premise is strengthened if the level of gene expression is related, in the predicted direction, to the phenotype in affected individuals. B. Causal pathway analysis for HHIP. SNPs that increase risk for low lung function and COPD increase lung tissue mRNA for HHIP AND increased lung tissue HHIP mRNA are associated with lower lung function and COPD. C. Causal pathway analysis for PCTH1. SNPs that increase risk for low lung function are associated with lower lung tissue mRNA for PTCH1 AND lower lung tissue PTCH1 mRNA is associated with lower lung function.
So what about this Hedgehog pathway could be important in the lung? The Hedgehog signaling pathway has been the subject of an excellent recent review,Citation19 and it is known that it coordinates embryonic lung development; however, recent evidence shows that it is also active during adulthood. Continued activation of the pathway maintains lung homeostasis and blockade is associated with an exaggerated mesenchymal proliferative response and aberrant repair of injured lungs.Citation20
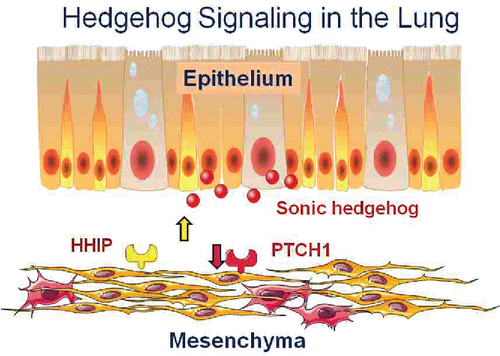
The ligand for the pathway, Sonic Hedgehog is secreted by the airway epithelium and acts on its receptor PTCH1 expressed on mesenchymal cells to activate hedgehog signaling. HHIP is also expressed on these cells and it can block the pathway. Thus, increased expression of the pathway blocker (HHIP) and decreased expression of the receptor are associated with low lung function suggesting that downregulation of this pathway in the adult lung is associated with lower lung function and maybe COPD. Conversely, activation of the pathway could be beneficial ().
Figure 11. Hedgehog signaling in the lung. The ligand Sonic Hedgehog is secreted by lung epithelial cells and acts on lung fibroblasts via its receptor PTCH1. HHIP negatively regulates the pathway by inhibiting the ligand. Increased HHIP and decreased PTCH1 are found in COPD lung tissue suggesting that decreased hedgehog signaling is a risk factor for COPD and, conversely, that stimulation of the pathway could be beneficial (See Reference Citation19).
Integrating genetics and genomics can also aid in understanding the role of biomarkers of disease. Biomarkers can be simple epiphenomena…markers that a disease process is ongoing or they may be involved in the causal pathway to disease. Take, for example, Surfactant Protein D. Serum levels of SFPTD are increased in COPD.Citation21 Does that mean SFPD is bad? Let me remind you that in Dr. Hobbs and Cho's GWAS for COPD SFTPD SNPs were associated with COPD.Citation9 To see if these SNPs influence surfactant expression, we performed a protein QTL analysis (pQTL). In a pQTL analysis, the effect of SNPs on the expression of a protein is measured instead of mRNA. Maen Obeidat and Don Sin recently measured serum SFTPD levels in over 4000 individuals in the Lung Health Study and found that the SNPs that were associated with risk for COPD and others in the region around the SFTPD gene on Chromosome 10 strongly affect serum SFPD levels (P < 1 × 10−90). When they examined the locus in more detail, they found 4 independent clusters of SNPs that influenced SFPD levels, some increasing and some decreasing surfactant protein in the serum. To understand the mechanism, they plotted the effect of the SNPs on the serum protein level versus the effect of the same SNPs on the lung mRNA for SFTPD. They found a strong relationship, some SNPs increasing and some decreasing SFPD protein and mRNA. This result indicates that the effect of the SNPs on the serum level of SFTPD is mediated by an effect on the mRNA. The SNPs are pQTLs because they are eQTLs.
Next, Obeidat and Sin plotted the effect of the SNPs on the risk for COPD in the international COPD Genetics Network cohortCitation9 versus the effect of the same SNPs on SFTPD serum levels and found that the SNPs that increased SFPD levels the most were associated with the lowest risk for COPD. The same was true in the Lung Health Study cohort; carriers of SNPs that increased SFTPD had a slower decline in lung function over the 5 years of that study. Therefore, although increased serum SFTPD is a marker of COPD, presumably secondary to lung damage, those individuals who make more SFTPD mRNA and protein are relatively protected from COPD.
The purpose of this review is to highlight the power of integrating genetic and genomic analysis for revealing potential causal pathways for respiratory disease. GWAS studies have identified many loci that increase risk for common respiratory diseases. Many of the genes and pathways were not considered a priori as candidates for susceptibility to lung diseases, which highlights our ignorance of the molecular mechanism underlying these disorders. Integrating genetics and genomics can reveal novel pathogenetic pathways that may serve as targets for disease modification. Importantly, these pathways may be involved in all, or most who have the disease, not just in carriers of the susceptibility alleles. Although such variants may be relatively rare and impart minor increases in relative risk for the disease in carriers of the alleles, the pathways that they help to identify may have substantial and generalized importance for disease pathogenesis.
References
- Wilmot KA, O'Flaherty M, Capewell S, Ford ES, Vaccarino V. Coronary heart disease mortality declines in the United States from 1979 through 2011: Evidence for stagnation in young adults, especially women. Circulation. 2015 Sep 15;132(11):997–1002.
- Khakban A, Sin DD, FitzGerald JM, et al. The projected epidemic of chronic obstructive pulmonary disease hospitalizations over the next 15 years. A population-based perspective. Am J Respir Crit Care Med. 2017 Feb 1;195(3):287–291.
- Benady S. The human and economic burden of COPD: a leading cause of hospital admission in Canada; 2010. Available from: http://www.respiratoryguidelines.ca/sites/all/files/CTS_COPD_report.pdf
- Sidney S, Quesenberry CP Jr, Jaffe MG, et al. Recent trends in cardiovascular mortality in the United States and public health goals. JAMA Cardiol. 2016 Aug 1;1(5):594–549.
- McDonough JE, Yuan R, Suzuki M, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med. 2011 Oct 27;365(17):1567–1575.
- Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004 Jun 24;350(26):2645–2653.
- Moffatt MF, Gut IG, Demenais F, et al. GABRIEL Consortium. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010 Sep 23;363(13):1211–1221.
- Hobbs BD, de Jong K, Lamontagne M, et al. Genetic loci associated with chronic obstructive pulmonary disease overlap with loci for lung function and pulmonary fibrosis. Nat Genet. 2017 Mar;49(3):426–432.
- Wain LV, Shrine N, Artigas MS, et al. Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential drugable targets. Nat Genet. 2017 Mar;49(3):416–425.
- Soler Artigas M, Loth DW, Wain LV, Gharib SA, Obeidat M, et al. Genome-wide association and large-scale follow up identifies 16 new loci influencing lung function. Nat Genet. 2011 Sep 25;43(11):1082–1090.
- Corvol H, Blackman SM, Boëlle PY, et al. Genome-wideassociationmeta-analysis identifies five modifier loci of lung disease severity incystic fibrosis. Nat Commun. 2015 Sep 29;6:8382.
- Noth I, Zhang Y, Ma SF, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: agenome-wide association study. Lancet Respir Med. 2013 Jun;1(4):309–317.
- Gauvreau GM, O'Byrne PM, Boulet LP, et al. Effects of an anti-TSLP antibody on allergen-induced asthmatic responses. N Engl J Med. 2014 May 29;370(22):2102–2110.
- Wenzel S, Ford L, Pearlman D, et al. Dupilumab in persistent asthma with elevated eosinophil levels. N Engl J Med. 2013 Jun 27;368(26):2455–2466.
- Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010 Apr 1;6(4):e1000888. doi: 10.1371/journal.pgen.1000888
- Maurano M.T., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012;337:1190–1195.
- Hao K, Bossé Y, Nickle DC, et al. Lung eQTLs to help reveal the molecular underpinnings of asthma. PLoS Genet. 2012;8(11):e1003029. doi: 10.1371/journal.pgen.1003029. Epub 2012 Nov 29.
- Obeidat M, Hao K, Bossé Y et al. Molecular mechanisms underlying variations in lung function: a systems genetics analysis. Lancet Resp Med. 015 Oct;3(10):782–795.
- Kugler MC, Joyner AL, Loomis CA, Munger JS. Sonic hedgehog signaling in the lung. From development to disease. Am J Respir Cell Mol Biol. 2015 Jan;52(1):1–13.
- Peng T, Frank DB, Kadzik RS, et al. Hedgehog actively maintains adult lung quiescence and regulates repair and regeneration. Nature. 2015 Oct 22;526(7574):578–582.
- Lomas DA, Silverman EK, Edwards LD, et al. Tal-Singer, and on behalf of the Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints Study Investigators. Serum surfactant protein D is steroid sensitive and associated with exacerbation of COPD. Eur Respir J. 2009;34:95–102.