
Abstract
Carbohydrates derivatives played an important and vital role in a large part to design new potential drug against various pathogens, in medicinal chemistry. Also, monosaccharide derivatives are very useful in biological chemistry because monosaccharide binding agents were shown to possess antiviral, antibacterial, and antifungal activity towards HIV, Dengue coronaviruses, and many members of the family Enterobacteriaceae, including strains of Escherichia coli, Salmonella typhimurium, Klebsiella pneumoniae, Shigella flexneri, and Enterobacter aerogenes. The present study was therefore interesting in identifying the binding affinity of a series of monosaccharides previously synthesized through their physicochemical and pharmacokinetic properties. These monosaccharides are derivatives of methyl-β-D-galactopyranoside (MGP) coupled to the protein deacetoxycephalosporin C synthase (DAOCS) of Streptomyces clavuligerus (1RXF). Molecular docking study against DAOCS proteins (PDB ID: 1RXF) was performed assess the antimicrobial activities of these derivatives along with antiviral prediction. The results of molecular docking studies showed that derivatives 8 and 10 both presented the highest score (-7.2 kcal/mol), reflecting their good binding affinity toward the active pocket of the pathogen S. clavuligerus (1RXF). They are found to be potent antimicrobial agents. These findings were further validated in dynamics (200 ns) milieu and intermolecular stability was evaluated via highly acceptable Molecular Mechanics Poisson-Boltzmann Surface Area (MMPB)/Molecular Mechanics/Generalized Born Surface Area (GBSA) binding free energies estimation methods. The dynamics of the complexes are highly stable with the strengthening of docked interactions as highlighted by MMPB/GBSA method (net binding energy < −40 kcal/mol). Density functional theory (DFT) calculations were carried out on the derivatives of MGP in order to determine the partial atomic charges, electronic energies, enthalpies, entropies and polarizability, at the B3LYP/3-21G level. The in silico ADMET prediction carried out provided access to the bioactivity and toxicity parameters which established that most of the different ligands designed are safe to use as drugs. This in silico study provided an overall view of the possible pharmacological and therapeutic profile of MGP derivatives. It therefore, paved the way for the discovery of promising next-generation drugs against pathogenic microbes.
1. Introduction
Carbohydrates are the most available biomolecules belonging to organic compounds class. They come from living organism’s kingdom. Around the world, scientists are investing in medicinal research to find more effective and safer antimicrobial agents for the treatment of diseases caused by pathogenic organisms. For a long time, carbohydrates remained an attractive research subject for scientists due to the important role played in the functioning of biological systems, namely the growth and proliferation of cells, communication between cells and the immune response (Nogueira, Parmanhan, Farias, & Corrêa, Citation2009). They are also the source of metabolic energy supply and responsible for the fine-tuning of cell-cell interactions and many other important biological processes (Sears & Wong, Citation1996; Werz, Citation2007). Other advantages of carbohydrates also pointed out are the numerous and diverse biological activities which they exert against pathogenic germs. In addition, recent advances in the treatment of infectious diseases have established that the use of carbohydrates could contain the effects of resistant races of microbes. In this case, the deep understanding of the antimicrobial activity mechanism is a better way which undoubtedly leads to synthesizing new antimicrobial compounds with improved and effective activity.
The literature survey revealed that most biologically active compounds contain aromatic, hetero-aromatic and acyl substituents (Alam et al., Citation2021a; Arifuzzaman, Islam, Rahman, Rahman, & Kawsar, Citation2018; Bulbul, Chowdhury, et al., Citation2021; Chowdhury, Bhuiyan, Ozeki, & Kawsar, Citation2016; Kawsar et al., Citation2018, Citation2015; Maowa, Asraful, et al., Citation2021; Rana, Ferdous, Hosen, & Kawsar, Citation2020). Benzene and its derivatives as well as nitrogen, sulfur and halogen used as substituents are known to improve the biological activity of the parent compound (Alam et al., Citation2021b; Devi et al., Citation2019; Rana et al., Citation2021; Yasmin, Amin, Hosen, & Sarkar, Citation2021b). It is also reported that by linking two active nuclei to each other, it is possible to obtain a new molecule with improved biological activity (Islam, Arifuzzaman, Rahman, Rahman, & Kawsar, Citation2019; Kawsar & Kumer, Citation2021).
Moreover, selective acylation as well as evaluation of microbial activities of carbohydrates revealed that in many cases the combination of two or more hetero-aromatic rings and acyl groups enhanced the biological activity of the parent nucleus (Kawsar, Faruk, Rahman, Fujii, & Ozeki, Citation2014; Kawsar et al., Citation2013; Misbah et al., Citation2020). Monosaccharide derivatives are known for their broad spectrum of biological activity against both Gram negative and positive organisms such as Escherichia coli, Bacillus subtilis, Salmonella typhi and Staphylococcus aureus (Islam et al., Citation2019; Kawsar et al., Citation2013; Citation2014; Misbah et al., Citation2020). In a recent study, some monosaccharide analogs were shown to exhibit remarkable inhibitory activity against cancer cell (Kawsar, Hosen, Fujii, Ozeki, & Thermochemical, Citation2020). Modifications to the hydroxyl group level in the nucleoside and monosaccharide structure have been shown very useful in the design of potent antiviral (M. Bulbul, Chowdhury, et al., Citation2021; Kawsar et al.,; Maowa, Asraful, et al., Citation2021; Yasmin et al., Citation2021a) and antimicrobial agents (Kawsar & Hossain, Citation2020; Kawsar et al., Citation2021). Thus, taking into account the observations and results mentioned above, we have chosen to substitute the hydrogen atom of the hydroxyl group of MGP with acyl substituents (including aromatic and hetero-aromatic group). Quantum chemistry methods are then used to determine some of electronic and thermochemical characteristics of the various resulting structures. Likewise, molecular docking studies were carried out to investigate the possible binding mechanism of MGP derivatives with DAOCS protein S. clavuligerus (PDB: 1RXF). The docking predictions on the binding mode of the compounds and interactions as not much reliable were thoroughly validated by dynamics stability in molecular dynamics simulations. Also, intermolecular strength of the complexes was confirmed through MMPB/GBSA analysis. On the other hand, and pharmacokinetic study using in silico ADMET prediction methods in order to demonstrate their bioavailability and toxicity, on the other hand. In general, the objective is to have, for MGP and derivatives, an in-depth knowledge of the in silico computational and pharmacokinetic properties.
2. Methodology
The following software’s were used in the present study:
Gaussian 09 (Frisch et al., Citation2016)
ii) AutoDock 4.2.6 (Forli et al., Citation2016),
iii) Swiss-Pdb 4.1.0 (Russo, Martin, & Hay, Citation1994),
iv) Python 3.8.2 (Lee, Yang, & Parr, Citation1988),
v) Discovery Studio 4.1 (Feng and Weissig, Citationn.d.),
vi) PyMOL 2.3 (DeLano, Citation2002),
vii) AVCPred (Guex & Peitsch, Citation1997),
viii) admetSAR server (Dallakyan & Olson, Citation2015).
2.1. Geometry optimization
In computational chemistry, quantum mechanical methods are widely used to calculate thermal, molecular orbital and molecular electrostatic properties (Berman et al., Citation2000). Geometry optimization of all the synthesized analogs is carried out using Gaussian 09 program (Frisch, Citation2009). Density functional theory (DFT) with Beck’s (B) (Becke, Citation1988) three parameter hybrid model, Lee, Yang and Parr’s (LYP) (Lee et al., Citation1988) correlation functional with 3-21 G basis set are employed to optimize and to predict their thermal and physicochemical properties. Parameters such as enthalpy, entropy, heat capacity, total energy, partial atomic charge and polarizability were calculated for all studied compounds.
2.2. Antiviral activities determination
Antiviral molecules (AVMs) are a special class of antimicrobial drugs used for the treatment of viral infections. They react by inhibiting the growth of the viral pathogen(s) inside the host cell. For the calculation of antiviral activity, we used the AVCPred program (Qureshi, Kaur, & Kumar, Citation2017) which displayed the inhibition percentage.
2.3. Preparation of protein and molecular docking
The molecular docking is one of the best and suitable approaches to identify the drug interactions with the protein DAOCS of Streptomyces clavuligerus. At this level, there is the blind docking method which consists of searching the whole surface of the protein molecule for binding sites.
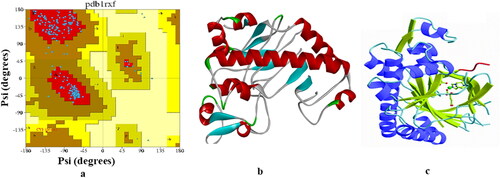
The 3 D structure of DAOCS (PDB ID. 1RXF) () was collected in pdb format from the protein data bank (Helen M Berman et al., Citation2000). All hetero-atoms and water molecules were took away by using PyMol (version 1.3) software packages (DeLano, Citation2002). Swiss-Pdb viewer software (version 4.1.0) was employed for energy minimization of the protein (Guex & Peitsch, Citation1997). Then optimized drugs were subjected from molecular docking study against S. clavuligerus protein (1RXF). In fine, molecular docking simulation was rendered by PyRx software (version 0.8) (Dallakyan & Olson, Citation2015) considering the protein as the macromolecule and the drug as the ligand. AutodockVina was used for docking analysis, and AutoDock Tools (ADT) of the MGL software package was used to convert pdb format into a Pdbqt format to input protein and ligands. The size of grid box in AutoDockVina was kept at 57.96, 48.57, and 47.22 Å for X, Y, Z directions respectively. Both macromolecule and ligand structures in Pdbqt format after the docking were used under Accelrys Discovery Studio (version 4.1) program to explore and visualize the result of docking and the search for non-bonding interactions between ligands and amino acid residues of receptor protein (Version, Citation2017). PDBsum program was also used to check the validation of the protein (PDB: 1RXF) following the scheme of Ramachandran (). It revealed that 93.20% of residues in the allowed region and no residues were missed.
Figure 1. (a): Ramachandran plot for the DAOCS (1RXF), (b, c): crystal structure and binding pocket of DAOCS (1RXF).
2.4. Validation protocol of docking
The biological validation of docking outcomes were investigated by extracting the co-crystallized ligand of the fungal protein (PDB ID: 1RXF) and re-docked it maintaining the same position. The ground state energy pose attained on re-docking and the co-crystallized ligands were superimposed, and its root mean square deviation (RMSD) score was calculated between these two superimposed ligands poses. To validate the docking procedure, The RMSD score range must be within the 2 Å (Onodera, Satou, & Hirota, Citation2007; Warren et al., Citation2006).
2.5. Molecular dynamics simulations
The selected complexes of compound 8 and 10 were prepared through LEAP module of AMBER20 (Case et al., Citation2020). First, the complexes were submerged in TIP3PBOX allowing a 12.0 Å padding distance between the protein and box edge. Subsequently, sufficient number of counter ions were used to neutralize charge of each system. The ANTECHAMBER program (Wang, Wang, Kollman, & Case, Citation2001) of the AMBER was employed to parameterize the compounds via GAFF force field (Wang, Wolf, Caldwell, Kollman, & Case, Citation2004), while FF14SB force field (Maier et al., Citation2015) was applied to the receptor protein. All atoms simulations of the complexes were performed after hydrogen atoms position optimization, water molecules optimization and entire systems minimization. The simulations were carried out with time scale of 2 fs and cutoff radius of 8.0 Å for non-bounded interactions. The long-range electrostatic interactions are calculated using particle-mesh Ewald method (Petersen, Citation1995) and hydrogen bonds were constrained via SHAKE algorithm (Kräutler, van Gunsteren, & Hünenberger, Citation2001). Systems heating was achieved in gradual phase from 0 to 300 K. Temperature control was accomplished through Langevin algorithm (Eisenmann & Stillfjord, Citation2021). Systems were then equilibrated for 100 ps at 1 atm pressure and 300 K. The same set of conditions were applied in 100 ns of simulations using PMEMD.CUDA from AMBER20. The simulations trajectories were investigated by CPPTRAJ module (Roe & Cheatham, Citation2013). The systems intermolecular docked conformation stability was examined by performing carbon alpha root mean square deviation (RMSD) (Kirchmair, Markt, Distinto, Wolber, & Langer, Citation2008) and root mean square fluctuation (RMSF) (Sahu & Pattanayak, Citation2019).
2.6. MMPB/GBSA binding free energies
The intermolecular interactions energy based on simulation trajectories was estimated using MMPB/GBSA method (Wang et al., Citation2019) by considering MMPBSA.py module (Miller et al., Citation2012) of the AMBER20. The binding energy analysis takes into account explicitly computed parameters involving gas phase energy, conformation entropy energy and solvation energy. Details of the MMPB/GBSA methodology applied herein are presented in Haq, Abro, Raza, Liedl, & Azam, Citation2017 (Haq et al., Citation2017). In total, 500 frames where the simulation trajectories were selected at regular time scale and used for the net binding free energy estimation.
2.7. Physicochemical pharmacokinetic analysis
The admetSAR 2.0 (http://lmmd.ecust.edu.cn/admetsar2/about) server was used to evaluate the pharmacokinetics and toxicity parameters related to drug absorption, metabolism and toxicity of the parent drug and its new designed analogs (Cheng et al., Citation2012). Using structure similarity search methods, ADMETSAR predicts the latest and most comprehensive manually accurate data for diverse chemicals associated with known ADME/T profiles. Molinspiration program (https://www.molinspiration.com/cgi-bin/properties) (Ali & Alam Khan, Citation2013) was employed to analyze the drug-likeness properties (Lipinski, Lombardo, Dominy, & Feeney, Citation1997) of lead compounds.
3. Results and discussion
In the present study, eleven derivatives of MGP obtained by proceeding by structural modification with aliphatic and aromatic chains (2–10) () were considered. Geometry optimization and antiviral activity prediction were performed to highlight the mode of their antimicrobial behavior. Observed activities were then rationalized by calculating parameters such as thermodynamic parameters, partial atomic charge, through molecular docking, with the combination in silico pharmacokinetic and drug-likeness properties.
Table 1. Structural views of MGP derivatives.
3.1. Structural designation of designed MGP derivatives
Atomic identification and structural variation of substituted MGP derivatives are displayed in . Different aliphatic (acetyl, butyryl, palmitoyl, myristoyl and steroyl) and aromatic (trityl, 3-chlorobenzoyl, cinnamoyl, and p-toluenesulfonyl) groups were subjected to substitute the hydrogen atom of MGP hydroxyl (-OH) group in order to observe the variation of biological activities.
3.2. FTIR spectrum
One of the most commonly employed methods that have been applied to detect organic and polymeric elements is Fourier transform infrared analysis (FTIR). This approach employs infrared light to inspect test materials and assess their chemical characteristics through using the FTIR analysis method. The FTIR system transmits infrared light of approximately 10,000 to 100 cm−1 through a material, with some energy consumed and some escaping through. Signals from analyzers normally range from 4000 cm−1 to 400 cm−1 which reflect the molecular fingerprint of the sample. The FTIR spectrum of MGP derivatives 2-10 represent in .
Figure 2. FTIR spectrum of the MGP derivatives (2-10).
3.3. Antiviral activity prediction
In order to check the antiviral mode of action of inserted aliphatic and aromatic groups, prediction of antiviral activities of studied carbohydrate derivatives (2-10) have been decided. Then predicted antiviral activities are compared to that of Azidothymidine (AZT, antiviral drug) using an antiviral application () (Qureshi et al., Citation2017).
Table 2. Predicted antiviral activities (% inhibition) of 2-10, and Azidothymidine (AZT).
The predicted antiviral activities revealed that the derivatives of MGP (2-10) possess potential antiviral efficacy when compared to their parent molecule. The aliphatic derivatives (2-6) and aromatic derivatives (7-10) exhibited promising scores in comparison with standard drugs Azidothymidine (AZT).
3.4. Physicochemical analysis
A simple alteration of chemical structure significantly influences the structural properties including thermal and molecular orbital properties. Spontaneity of a reaction and stability of a product can be known from calculating its free energy and enthalpy values respectively (Cohen & Benson, Citation1993). Highly negative values are more eligible for gaining thermal stability. In drug design, hydrogen bond formation and non-bonded interactions are also influenced by dipole moment.
Electronic energy is a significant criterion to represent the interaction of binding partners, where negative value favors spontaneous binding and interaction. In the present study, the calculated properties values of all the MGP derivatives are greater negative than those of the parent MGP. Hence, these observations indicated that the attachment of ester group could improve the interaction and binding of these molecules with different microbial enzymes. As shown in , physicochemical data revealed that, among all the synthesized derivatives, only compound (6) with the highest molecular weight showed the highest value for polarizability (524.901a.u), Heat capacity (309.642 cal/mol-kelvin) and Entropy (425.076 cal/mol-kelvin). All the derivatives with aromatic ring as substituent displayed higher score comparatively to the aliphatic one. Finally, this discussion proves that the substitution carried out at the level of hydroxyl (-OH) groups of MGP increases significantly its thermodynamic properties which indicate the inherent stability of the synthesized derivatives. Moreover, presence of bulky acylating groups also suggesting the possible improvement of these physicochemical parameters.
Table 3. Electronic energy (Hartree), entropy (cal/mol-kelvin), heat capacity (cal/mol-kelvin) and polarizability (a.u.) of MGP derivatives.
3.5. Atomic partial charge analysis: NBO
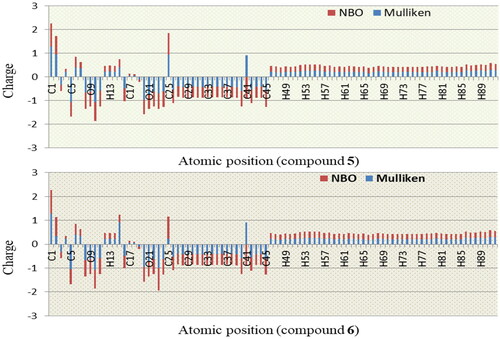
The partial atomic charge is essential for such purposes in molecular computations as a simplified representation of global charge distribution in a molecule and predicting its conformational behavior. NBO analysis is carried out in order to know the charge distribution and the intrinsic property of the interactions in the designed structure. Polarity of chemical bonds often influences the structure and reactivity of a molecule (Heinz & Suter, Citation2004). The molecular dipole moment is a vector, which does not clearly define the polarity of the molecule. Different methods have been proposed for assigning partial charges to the interacting atoms within a molecule. The approaches of Mulliken Population Analysis and Natural Bond Orbital were employed to compute the partial charges of all drug interacting atoms (Gross, Seybold, & Hadad, Citation2002). They are the most used population analysis methods. Dipole moment and molecular polarizability are related to atomic charges (Mulliken, Citation1955). Here, all the hydrogen atoms showed positive charge in both methods and other electronegative atom (O), negative charge in both methods as expected (). In derivative 5 (C-1, C-2 and C-25) showed greater positive charges. These greater positive charges are found due to the presence of higher electronegative element like oxygen (O-9, O-21, and O-33), and hydrogen H-7 which exhibit the higher positive value than the other hydrogen because of oxygen atom of hydroxyl group. Similarly, the carbon atoms identified in the derivatives 6 (C-1, C-6, and C-25), 7 (C-1, C-25), and 8 (C-5, C-14) displayed positive charge in both methods due to the presence of the oxygen atom of carbonyl group. In all derivatives, the carbon (C-25) showed the highest positive charge in all the other drugs. This fact is due to the presence of greater electronegative atom (N, Cl and O). As derivatives (2-6) contain aliphatic substituents and particularly derivatives 5 and 6 possess the longest carbon chain. It was found that these two compounds showed results that are improved. Finally, we depicted derivative 5 and 6 () from both aliphatic and aromatic series, as these compounds exhibited clear graphical view while the rest of the derivatives gave slightly scribble view.
Figure 3. Atomic partial charges of derivatives 5 and 6.
3.6. Validation study
The identification of the molecular docking algorithms to confirm the conformation of the protein-bound ligand, performed by re-docking of the co-crystallized ligand pose which revealed for the validation effectiveness of the docking results. The superimposed pose between the docked ligand conformation and the co-crystallized ligand conformation revealed the RMSD value is < 2Ǻ. The complex was then observed to interact with the all similar amino acid residues compared to the ones reported in the current study. The large symmetric structure can be interacted in the active site of protein during docking, as the case in this study; the RMSD score would be at a very high level. Again, the small molecules can attain low RMSD score easily even when imposed randomly. Some reported works have found a new strategy for the efficacy of docking poses on the basis of visual realization. The outcomes of this visual observation showed the same interactions as in the experimental binding mode, as found in docking.
3.7. Molecular docking simulation
Molecular docking is a type of bioinformatics modeling which deals with the interaction of two or more molecules to give the stable adduct. In and Table S1 the derivatives of MGP (2-5, 8-10) showed excellent docking performance and the chosen proteins were discovered to communicate with significant amino acids. We docked totally 9 complexes having better antimicrobial activities for one protein with eight MGP derivatives. We choose the deacetoxycephalosporin c synthase (DAOCS) protein (PDB: 1RXF) from S. clavuligerus bacteria and all designed derivatives were docked into the same binding pocket of DAOCS using similar optimized docking conditions to investigate their binding mode. The outcomes of the docking analysis showed that obtained binding affinities are ranging from −5.2 to −7.2 kcal/mol for all derivatives and parent compound. As shown in , all MGP derivatives (2-6 and 8-10) displayed higher binding affinity than parent MGP. These results indicated that insertion of a long carbon chain/aromatic ring allow the molecule to improve binding affinity, while addition of hetero groups like -Cl and –CH3 made some fluctuations in binding affinities. However, modification with halogenated aromatic rings increased the binding affinity. The docked pose clearly showed that the drugs molecules bind within the active site of the DAOCS macromolecular structure (). Comparatively the aromatic analogs displayed better binding score than aliphatic analogs. The interactions between inhibitors and bordering residues of DAOCS are illustrated in the 2 D schematics. These pictures were obtained by importing them from docking output to the Discovery Studio Visualizer and lig-plot ( and and S1-S2). Obtained results show that amino acids participate in the pattern of interactions between the ligand and protein with an important contribution to the total energy of interaction.
Figure 4. Docking conformation of derivative (8) at inhibition bounding site of 1RXF (a) and Docking conformation of derivative (10) at inhibition bounding site of 1RXF (b).
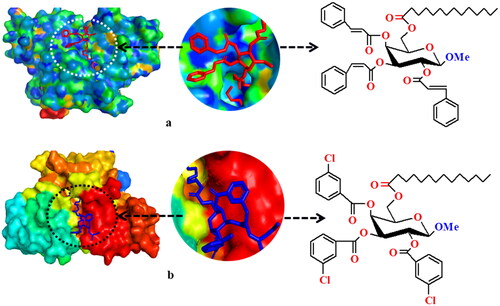
Figure 5. (a): Lig-plot and, (b): Multiple sequence alignment of closest homologs of DAOCS (PDB: 1RXF).
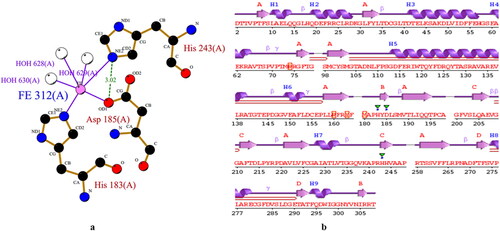
Figure 6. Non-bonding interactions of derivatives (3, 4, 8, and 10) with the amino acid residues of 1RXF generated by Discovery Studio.
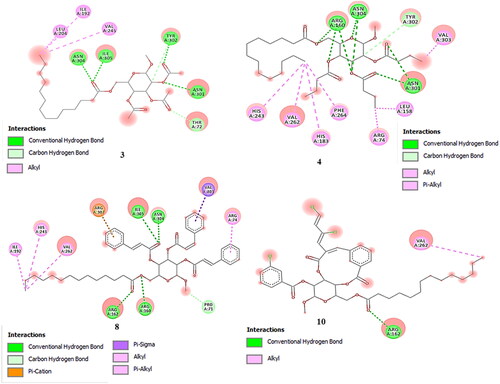
Table 4. Binding energy and non-bonding interaction data of MGP derivatives against S. clavuligerus 1RXF.
Most of these interactions include hydrophobic contacts, Van der Waals interactions, hydrogen bonds, electrostatic, carbonyl, and one specific atom-aromatic ring provide insight help to understand molecular recognition. As shown in aromatic derivatives (2-5) except compound 6; display slight rising of binding score (-6.0, −6.4 − 6.3 and-6.3 kcal/mol) by increasing of chain C2-C16 length. Most prominent hydrogen bonded binding sites observed for these derivatives were Arg160, Arg266, Arg306, Asn301, Asn304, Tyr184, Tyr302, Thr 72, Trp297 and Ile305 (conventional hydrogen bond) and Tyr302, Thr 72 (carbon hydrogen bond). Moreover, hydrophobic interactions (alkyl and pi-alkyl) found for the residues Ile305, Ile192, His183, His243, Phe264, Arg242, Arg302, Val245, Val262, Lys239 and Val303. From displayed data in below it is clear that; the binding sites were almost similar for aliphatic derivatives where Trp297, Thr72 and Asn304 showed shortest distances which are 1.886 Å, 1.909 Å and 1.929 Å respectively.
Table 5. MMPB/GBSA energy parameters estimated for selected best docked complexes. The energy values are expressed in kcal/mol.
Among aliphatic derivatives, compounds 6 displays binding score which is fluctuating more than other aliphatic analogs with new binding sites. Aromatic derivative (8-10) displayed higher fluctuating binding score (-7.2, −6.9 and −7.2 kcal/mol) compared to aliphatic derivatives. These derivatives are also interacting with catalytic active site of the bacterial protein through hydrogen bond (Arg160, Arg162, Arg306, Asn304, Ile305, Pro71, Tyr184 Asp284 Thr308, and Phe283), hydrophobic bond (Arg307, Arg74, Ile192, His243, Val262 and Lys239) and electrostatic bond (Arg307, Arg303). Pi-cation and pi-sigma interactions are found only at the level of aromatic derivatives.
So, these outcomes show clearly that the fact of having high electron density, aromatic substituents can easily increase the binding skill as well as the antibacterial power of MGP derivatives. According to Phe264, all the derivatives displayed the maximum π-interactions with the His243, His183, Arg306And Arg307 denoting the tight binding with the active site. Reports suggest that phenylalanine and histidine residues are considered as the principal component of the π-alkyl, π-sigma and π-cation. They are responsible for the accessibility of small molecules to the active site. Binding energy and binding mode were improved in derivatives (2-5 and 8-10) because of significant hydrogen bonding. It was observed that the alterations of the -OH group in MGP exalted the π–π interactions with the amino acid chain on the binding site, whereas their polarity improvement resulted in the formation of hydrogen bond interactions.
The most significant H-bonds were obtained for the derivative (3 and 9), forming with identical residues such as Arg306, Arg160, Tyr184, Tyr302, Asn304, Phe283, Asp284, Thr72 and Ile305. Hydrogen-bonds execute a vital function in shaping the specificity of ligand binding with the receptor, drug design in chemical and biological processes, molecular recognition and biological activity (Perlstein, Citation2001).
Hydrogen bond surface and hydrophobic surface of derivative (3) is represented in . We realize that the analyzed MGP derivatives bind within the active site of the DAOCS protein of S. clavuligerus, which is inevitable to prevent the protein from its infections. The calculated binding affinities varied from −5.2 to −7.2 kcal/mol which suggesting that the molecules can spontaneously interact within the binding site of bacterial protein. The docking findings therefore indicate that the derivatives 8 and 10 may possess excellent effectiveness for S. clavuligerus.
Figure 7. Hydrogen bond surface (a) and hydrophobic surface (b) of 1RXF with derivative (3).
3.8. Molecular dynamics simulations studies
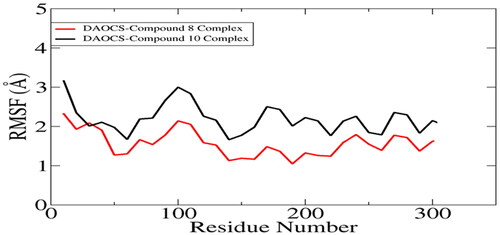
Molecular dynamics simulations were conducted in order to decipher selected compounds docked conformation stability with the proteins as well as to determine the strength of interactions between the molecules. It can be seen in , that both selected docked complexes are depicting stable dynamics behavior: the DAOCS-compound 8 complex is relatively more stable than that of compound 10. The latter system experienced some deviations between times 10 ns to 65 ns however, received considerable stable towards simulation end time. RMSD for both systems are around 2.5 Å. The RMSD was further extended to validate stability of systems (). DAOCS-compound 8 complex was unveiled to gain better stability compared to DAOCS-compound 10 in the last 100 ns. Nevertheless, both systems revealed good in-termolecular binding conformation and interaction throughout the length of simulation time. Next, RMSF was calculated, which concluded that majority of the receptor residues are within acceptable flexibility range with RMSF value < 2 Å (). The terminal regions of the proteins showed some fluctuations though that were to minor to effect overall systems stability.
Figure 8. RMSD analysis of selected docked complexes based on carbon alpha atoms.
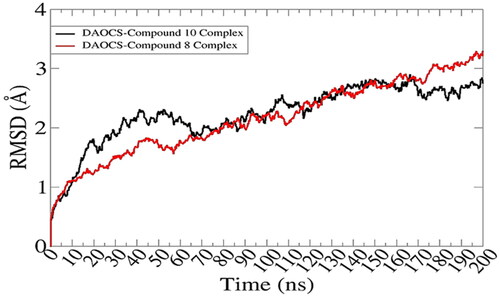
Figure 9. RMSF analysis of selected docked complexes based on carbon alpha atoms.
3.9. MMPB/GBSA binding free energy analysis
The MMPB/GBSA analysis was performed using snapshots extracted from simulation trajectories to get validation on the docking predictions. Likewise, the docking studies the binding free energy analysis by MMPB/GBSA method also reported both the complexes as excellent stable complexes as demonstrated by negative net binding free energy that is −47.5 kcal/mol for DAOCS-compound 8 complex in MM/GBSA and −41.49 kcal/mol in MM/PBSA. While for DAOCS-compound 10 complex, the net MM/GBSA binding free energy is −46.71 kcal/mol and MM/PBSA binding free energy is −39.24 kcal/mol. These energy values are four times more than that predicted by the docking study which may be the fact that as multiple dynamics conformations of the complexes are selected compared to just one docked conformation in docking studies. Also, it indicates that as simulation proceeds the binding affinity of the compounds get more strengthen. Details of the different energy parameters contribution to the net binding energy values is given in .
3.10. Pharmacokinetic and drug-likeness properties
Molecular parameters like membrane permeability and bioavailability of lead molecules depend on some fundamental features of molecules like partition coefficient (logP), molecular weight (MW), and number of hydrogen bond acceptors/donors that is associated with Lipinski ‘‘rule of five’’. exhibits derivatives (2-4) are in agreement with the Lipinski rule of five indicating the good bioavailability of MGP derivatives, whereas derivatives (5-10) don’t satisfy half of Lipinski’s rules.
Table 6. Prediction Drug likeness properties of MGP derivatives.
Derivatives with low molecular weight are easily absorbed, diffused and transported as compared to high molecular weight ones. As molecular weight increases except certain limit, the bulkiness of the molecules is also increasing. TPSA (Topological Polar Surface Area) is very important physicochemical property of molecule that provides the information about polarity of compounds. This parameter was screened for analyzing drug transport properties. Polar surface area is the sum of all polar atoms mainly oxygen and nitrogen including attached hydrogen. The drug-likeness score of lead derivatives is predicted with combination of GPCR, ion channel modulator, kinase inhibitor, nuclear receptor ligands, protease inhibitor, and enzyme inhibitor, which has been employed to identify the efficacy of molecules. When the bioactivity score is larger that means that the probability of the specific molecule to be active is higher. If bioactivity score of molecules is greater than 0.00, that molecule is qualified to be promising biological activities. When the scores are ranging from −0.50 to 0.00, they are considered to be moderately active. Finally, if bioactivity scores are less than −0.50 it is presumed to be inactive. The bioactivity scores of all designed MGP derivatives are displayed in . The obtained values of drug likeness scores exhibited that derivatives (2-4 and 8-10) followed the promising drug-likeness.
Table 7. Determination of drug likeness score of MGP derivatives through molinspiration cheminformatics online server.
The bioactivity score gives the information about the binding cascade of the drugs that is applied for the development of a new functional drug with increased binding selectivity profile and less undesirable effects. All the selected MGP derivatives were evaluated to toxicity profile and given in above.
Table 8. Pharmacokinetic parameters of MGP derivatives.
3.11. Calculation of QSAR and pIC50
The QSAR study is a computational modeling pathway for investigating correlations among the structural features of chemical substances and biological activity. To calculate the QSAR and pIC50 value, we have used web tool called Chemdesk and takes standard value including Chiv5, MRVSA9, PEOEVSA5, GATSv4 etc. After that, multiple linear regression (MLR) equation is utilized to obtained the QSAR and pIC50 value (84).Our compounds has been shown and meet all the criteria and different QSAR and pIC50.The range of the QSAR and pIC50 Lowest value been obtained 4.13 where the highest value has obtained 7.72 ().
Table 9. QSAR Data of MGP derivatives.
4. Conclusions
In this investigation, the inherent characteristic of stability and biochemical behavior of MGP and its derivatives are studied for suggesting new antimicrobial compounds with improved activity. Important properties of biological chemistry and chemical reactivity such as antiviral activity, atomic partial charges and physicochemical properties were evaluated. They indicated that these derivatives are good candidate molecules for the development of drugs. All the designed MGP derivatives have improved physicochemical score than parent MGP. It is resulting that modified compounds are more reactive than parent drug. Insertion of various aliphatic and aromatic groups in MGP structure can significantly improve their biological activity. Antiviral prediction of MGP derivatives 2–10 revealed their promising potential activity against various pathogens. These observations were rationalized by molecular docking that pointed out promising antimicrobial effectiveness of MGP derivatives. Many of these analogs showed remarkable binding interactions and binding energy with DAOCS protein of S. clavuligerus. MGP derivatives (3-4, 8, and 10) have shown in silico potent ability to fight against S. clavuligerus. The selected complexes of derivatives 8 and 10 are also dynamically stable and interaction between the compound and receptor protein is dominated by van der Waals and electrostatic interactions. Finally, these derivatives were analyzed for their pharmacokinetic and drug-likeness properties which allowed understanding that the combination of toxicity prediction, bioactivity prediction, and drug-likeness had promising results. These promising results are obtained because most of designed molecules have improved kinetic parameters and all rules of drug-likeness are satisfied. In terms of biological activity, the results obtained were interesting result. Finally, this research may be useful to understand the chemical, thermal, physicochemical, biological and pharmacokinetic properties of MGP derivatives.
Data availability statement
Data are available in this article and supplementary materials.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Alam, A., Hosen, M. A., Hosen, A., Fujii, Y., Ozeki, Y., & Kawsar, S. (2021a). Cómo citar Buscar synthesis, characterization, and molecular docking against a receptor protein FimH of Escherichia coli (4XO8) of thymidine derivatives Journal of the Mexican Chemical Society, vol. 65, no. 2, 2021 Sociedad Química de México AC. Journal of the Mexican Chemical Society, 65(2), 256–276. doi:10.29356/jmcs.v65i2.1464
- Alam, A., Hosen, M. A., Islam, M., Ferdous, J., Fujii, Y., Ozeki, Y., & Kawsar, S. (2021b). Synthesis, antibacterial and cytotoxicity assessment of modified uridine molecules. Current Advances in Chemistry and Biochemistry, 6, 114–129.
- Ali, M. M., & Alam Khan, S. (2013). Virtual screening of molecular properties and bioactivity score of boswellic acid derivatives in search of potent anti-inflammatory lead molecule. International Journal of Interdisciplinary and Multidisciplinary Studies, 1, 8–12.
- Arifuzzaman, M., Islam, M. M., Rahman, M. M., Rahman, M. A., & Kawsar, S. (2018). An efficient approach to the synthesis of thymidine derivatives containing various acyl groups: Characterization and antibacterial activities. ACTA Pharmaceutica Sciencia, 56(4), 7. doi:10.23893/1307-2080.APS.05622
- Becke, A. D. (1988). Density-functional exchange-energy approximation with correct asymptotic behavior. Physical Review. A, General Physics, 38(6), 3098–3100. doi:10.1103/physreva.38.3098
- Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., weissig, H., … Bourne, P. E. (2000). The protein data bank. Nucleic Acids Research, 28(1), 235–242. doi:10.1093/nar/28.1.235
- Bulbul, M., Chowdhury, T. S., Misbah, M., Ferdous, J., Dey, S., Hasan, I., … Kawsar, S. (2021). Synthesis of new series of pyrimidine nucleoside derivatives bearing the acyl moieties as potential antimicrobial agents. Pharmacia, 68(1), 23–34. doi:10.3897/pharmacia.68.e56543
- Bulbul, M., Hosen, M., Ferdous, J., Chowdhury, T., Misbah, M., & Kawsar, S. (2021). DFT study, physicochemical, molecular docking, and ADMET predictions of some modified uridine derivatives. International Journal of New Chemistry, 8, 88–110.
- Case, D. A., Belfon, K., Ben-Shalom, I., Brozell, S. R., Cerutti, D., Cheatham, T., … Giambasu, G. (2020). Amber 2020. University of California, San Francisco.
- Cheng, F., Li, W., Zhou, Y., Shen, J., Wu, Z., Liu, G., … Tang, Y. (2012). admetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. Journal of Chemical Information and Modeling, 52(11), 3099–3105.
- Chowdhury, S., Bhuiyan, M., Ozeki, Y., & Kawsar, S. (2016). Simple and rapid synthesis of some nucleoside derivatives: Structural and spectral characterization. Current Chemistry Letters, 5, 83–92. doi:10.5267/j.ccl.2015.12.001
- Cohen, N., & Benson, S. W. (1993). Estimation of heats of formation of organic compounds by additivity methods. Chemical Reviews, 93(7), 2419–2438. doi:10.1021/cr00023a005
- Dallakyan, S., & Olson, A. J. (2015). Small-molecule library screening by docking with PyRx. In Chemical biology (pp. 243–250). Springer.
- DeLano, W. L. (2002). The PyMOL molecular graphics system. http://www.pymol.org.
- Devi, S. R., Jesmin, S., Rahman, M., Manchur, M. A., Fujii, Y., Ozeki, Y., & Kawsar, S. (2019). Microbial efficacy and two step synthesis of uridine derivatives with spectral characterization. ACTA Pharmaceutica Sciencia, 57(1), 47. doi:10.23893/1307-2080.APS.05704
- Eisenmann, M., & Stillfjord, T. (2021). Sub-linear convergence of a tamed stochastic gradient descent method in Hilbert space. arXiv preprint arXiv:2106.09286. https://doi.org/10.48550/arXiv.2106.09286
- Feng, Z., & Weissig, G. (n.d). IN Shindyalov PE Bourne (2000). Nucleic Acids Research, 28, 235.
- Forli, S., Huey, R., Pique, M. E., Sanner, M. F., Goodsell, D. S., & Olson, A. J. (2016). Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nature Protocols, 11(5), 905–919.
- Frisch, M. J. (2009). Gaussian 09, Revision D. 01. Wallingford, CT: Gaussian, Inc.
- Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, A., Cheeseman, J. R., … Petersson, G. A. (2016). Gaussian 09, Revision A.02. Wallingford, CT: Gaussian Inc.
- Gross, K. C., Seybold, P. G., & Hadad, C. M. (2002). Comparison of different atomic charge schemes for predicting pKa variations in substituted anilines and phenols. International Journal of Quantum Chemistry, 90(1), 445–458. doi:10.1002/qua.10108
- Guex, N., & Peitsch, M. C. (1997). SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis, 18(15), 2714–2723. doi:10.1002/elps.1150181505
- Haq, F. U., Abro, A., Raza, S., Liedl, K. R., & Azam, S. S. (2017). Molecular dynamics simulation studies of novel β-lactamase inhibitor. Journal of Molecular Graphics & Modelling, 74, 143–152. doi:10.1016/j.jmgm.2017.03.002
- Heinz, H., & Suter, U. W. (2004). Atomic charges for classical simulations of polar systems. The Journal of Physical Chemistry B, 108(47), 18341–18352. doi:10.1021/jp048142t
- Islam, M., Arifuzzaman, A., Rahman, M., Rahman, M. A., & Kawsar, S. (2019). Novel Methyl 4, 6-O-benzylidene-α-D-glucopyranoside derivatives: Synthesis, structural characterization and evaluation of antibacterial activities. Hacettepe Journal of Biology and Chemistry, 47, 153–164. doi:10.15671/hjbc.622038
- Kawsar, S., Ara, H. A., Uddin, S. A., Hossain, M. K., Chowdhury, S. A., Sanaullah, A., … Fujii, Y. (2015). Chemically modified uridine molecules incorporating acyl residues to enhance antibacterial and cytotoxic activities. International Journal of Organic Chemistry, 5(4), 232–245. doi:10.4236/ijoc.2015.54023
- Kawsar, S., Faruk, M. O., Rahman, M. S., Fujii, Y., & Ozeki, Y. (2014). Regioselective synthesis, characterization, and antimicrobial activities of some new monosaccharide derivatives. Scientia Pharmaceutica, 82(1), 1–20. doi:10.3797/scipharm.1308-03
- Kawsar, S., Hasan, T., Chowdhury, S. A., Islam, M. M., Hossain, M. K., & Manchur, M. A. (2013). Synthesis, spectroscopic characterization and in vitro antibacterial screening of some D-glucose derivatives. International Journal of Pure Applied Chemistry, 8, 125–135.
- Kawsar, S., Hosen, M. A., Chowdhury, T. S., Rana, K. M., Fujii, Y., & Ozeki, Y. (2021). Thermochemical, PASS, molecular docking, drug-likeness and in silico ADMET prediction of cytidine derivatives against HIV-1 reverse transcriptase. Revista de Chimie, 72(3), 159–178.
- Kawsar, S., Hosen, M. A., Fujii, Y., Ozeki, Y., & Thermochemical, D. (2020). molecular docking and pharmacokinetic studies of methyl β-D-galactopyranoside esters. SDRP Journal of Computational Chemistry & Molecular Modelling, 4(4), 452–462. doi:10.25177/JCCMM.4.4.RA.10663
- Kawsar, S., & Hossain, M. A. (2020). An optimization and pharmacokinetic studies of some thymidine derivatives. Turkish Computational and Theoretical Chemistry, 4(2), 59–66. doi:10.33435/tcandtc.718807
- Kawsar, S., Islam, M., Jesmin, S., Manchur, M. A., Hasan, I., & Rajia, S. (2018). Evaluation of the antimicrobial activity and cytotoxic effect of some uridine derivatives. International Journal of Bioscience, 12, 211–219.
- Kawsar, S., & Kumer, A. (2021). Computational investigation of methyl α-D-glucopyranoside derivatives as inhibitor against bacteria, fungi and COVID-19 (SARS-2). Journal of the Chilean Chemical Society, 66(2), 5206–5214. doi:10.4067/S0717-97072021000205206
- Kirchmair, J., Markt, P., Distinto, S., Wolber, G., & Langer, T. (2008). Evaluation of the performance of 3D virtual screening protocols: RMSD comparisons, enrichment assessments, and decoy selection-what can we learn from earlier mistakes? Journal of Computer-Aided Molecular Design, 22(3–4), 213–228. doi:10.1007/s10822-007-9163-6
- Kräutler, V., van Gunsteren, W. F., & Hünenberger, P. H. (2001). A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. Journal of Computational Chemistry, 22(5), 501–508. doi:10.1002/1096-987X(20010415)22:5<501::AID-JCC1021>3.0.CO;2-V
- Lee, C., Yang, W., & Parr, R. G. (1988). Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Physical Review. B, Condensed Matter, 37(2), 785–789. doi:10.1103/physrevb.37.785
- Lipinski, C. A., Lombardo, F., Dominy, B. W., & Feeney, P. J. (1997). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews, 23(1–3), 3–25. doi:10.1016/S0169-409X(96)00423-1
- Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., & Simmerling, C. (2015). ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. Journal of Chemical Theory and Computation, 11(8), 3696–3713. doi:10.1021/acs.jctc.5b00255
- Maowa, J., Asraful, A., Kazi, M., Sujan, D., Hosen, A., Fujii, Y., … Kawsar, S. (2021). Synthesis, characterization, synergistic antimicrobial properties and molecular docking of sugar modified uridine derivatives. Ovidius University Annals of Chemistry, 32(1), 6–21. doi:10.2478/auoc-2021-0002
- Maowa, J., Hosen, M. A., Alam, A., Rana, K. M., Fujii, Y., Ozeki, Y., & Kawsar, S. (2021). Pharmacokinetics and molecular docking studies of uridine derivatives as SARS-COV-2 Mpro inhibitors. Physical Chemistry Research, 9, 385–412.
- Miller III, B. R., McGee Jr, T. D., Swails, J. M., Homeyer, N., Gohlke, H., & Roitberg, A. E. (2012). MMPBSA.py: An efficient program for end-state free energy calculations. Journal of Chemical Theory and Computation, 8(9), 3314–3321. doi:10.1021/ct300418h
- Misbah, M., Ferdous, J., Bulbul, M., Chowdhury, T. S., Dey, S., Hasan, I., & Kawsar, S. (2020). Evaluation of MIC, MBC, MFC and anticancer activities of acylated methyl β-D-galactopyranoside esters. International Journal of Bioscience, 16, 299–309.
- Mulliken, R. S. (1955). Electronic population analysis on LCAO–MO molecular wave functions. II. Overlap populations, bond orders, and covalent bond energies. The Journal of Chemical Physics, 23(10), 1841–1846. doi:10.1063/1.1740589
- Nogueira, C. M., Parmanhan, B. R., Farias, P. P., & Corrêa, A. G. (2009). A importância crescente dos carboidratos em química medicinal. Revista Virtual de Química, 1(2), 149–159. doi:10.5935/1984-6835.20090017
- Onodera, K., Satou, K., & Hirota, H. (2007). Evaluations of molecular docking programs for virtual screening. Journal of Chemical Information and Modeling, 47(4), 1609–1618. doi:10.1021/ci7000378
- Perlstein, J. (2001). The weak hydrogen bond in structural chemistry and biology (International Union of Crystallography, Monographs on Crystallography, 9) By Gautam R. Desiraju (University of Hyderabad) and Thomas Steiner (Freie Universität Berlin). Oxford University Press.
- Petersen, H. G. (1995). Accuracy and efficiency of the particle mesh Ewald method. The Journal of Chemical Physics, 103(9), 3668–3679. doi:10.1063/1.470043
- Qureshi, A., Kaur, G., & Kumar, M. (2017). AVCpred: an integrated web server for prediction and design of antiviral compounds. Chemical Biology & Drug Design, 89(1), 74–83. doi:10.1111/cbdd.12834
- Rana, K. M., Ferdous, J., Hosen, A., & Kawsar, S. (2020). Ribose moieties acylation and characterization of some cytidine analogs. Journal of Siberian Federal University. Chemistry, 13, 465–478. doi:10.17516/1998-2836-0199
- Rana, K. M., Maowa, J., Alam, A., Dey, S., Hosen, A., Hasan, I., … Kawsar, S. (2021). In silico DFT study, molecular docking, and ADMET predictions of cytidine analogs with antimicrobial and anticancer properties. In Silico Pharmacology, 9(1), 1–24. doi:10.1007/s40203-021-00102-0
- Roe, D. R., & Cheatham, T. E. III. (2013). PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. Journal of Chemical Theory and Computation, 9(7), 3084–3095. doi:10.1021/ct400341p
- Russo, T. V., Martin, R. L., & Hay, P. J. (1994). Density functional calculations on first‐row transition metals. The Journal of Chemical Physics, 101(9), 7729–7737. doi:10.1063/1.468265
- Sahu, S. N., & Pattanayak, S. K. (2019). Molecular docking and molecular dynamics simulation studies on PLCE1 encoded protein. Journal of Molecular Structure, 1198, 126936. doi:10.1016/j.molstruc.2019.126936
- Sears, P., & Wong, C.-H. (1996). Intervention of carbohydrate recognition by proteins and nucleic acids. Proceedings of the National Academy of Sciences of the United States of America, 93(22), 12086–12093. doi:10.1073/pnas.93.22.12086
- Version, A. D. S. (2017). 4.0. San Diego: Accelrys.
- Wang, E., Sun, H., Wang, J., Wang, Z., Liu, H., Zhang, J., & Hou, T. (2019). End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chemical Reviews, 119(16), 9478–9508. doi:10.1021/acs.chemrev.9b00055
- Wang, J., Wang, W., Kollman, P. A., & Case, D. A. (2001). Antechamber: An accessory software package for molecular mechanical calculations. Journal of the American Chemical Society, 222, U403.
- Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A., & Case, D. A. (2004). Development and testing of a general amber force field. Journal of Computational Chemistry, 25(9), 1157–1174. doi:10.1002/jcc.20035
- Warren, G. L., Andrews, C. W., Capelli, A.-M., Clarke, B., LaLonde, J., Lambert, M. H., … Head, M. S. (2006). A critical assessment of docking programs and scoring functions. Journal of Medicinal Chemistry, 49(20), 5912–5931. doi:10.1021/jm050362n
- Werz, D. B. (2007). Chemistry-A European J 2005; 11: 3194–3206.(c) Seeberger PH, Werz DB. Nature, 446, 1046–1051. [Mismatch
- Yasmin, F., Amin, M. R., Hosen, M. A., Bulbul, M., Dey, S., & Kawsar, S. (2021a). Monosaccharide derivatives: Synthesis, antimicrobial, PASS, antiviral and Molecular docking studies against SARS-COV-2 Mpro inhibitors. Cellulose Chemistry and Technology, 55(5–6), 477–499. doi:10.35812/CelluloseChemTechnol.2021.55.44
- Yasmin, F., Amin, M. R., Hosen, A., & Sarkar, M. K. (2021b). Bromobenzoylation of Methyl α-D-Mannopyranoside: Synthesis and spectral characterization. Journal of Siberian Federal University. Chemistry, 14, 171–183. doi:10.17516/1998-2836-0226