
Abstract
A series of phthalazine derivatives have been synthesized using green protocol methods under microwave irradiation. This involved subjecting chlorophthalazine to microwave irradiation and employing various reagents, including hydrazine derivatives, primary and secondary amines, and active methylene, to generate the novel phthalazine derivatives. Subsequently, a comparison was made between the yield percentage and reaction time of the resulting products obtained from conventional methods and those obtained through microwave irradiation. The structures of all new compounds were confirmed by physical properties and spectral analysis. The phthalazine compounds demonstrated significant anticancer activity against three cancer cell lines and showed promising selectivity when compared with doxorubicin. Compound 6 exhibited the best anticancer activity, with IC50 1.739 µM, 0.384 µM, and 1.52 µM against MCF7, HCT116, and HepG2 cell lines, respectively. The docking results confirmed that the drugs exert their activity by inhibiting PARP-1. The binding scores were in accordance with the experimental IC50, with compound 6 exhibiting the best binding score and retaining the essential binding interactions with the active site. The results of our computational research have revealed that our phthalazine compounds possess a unique structure that effectively inhibits PARP-1.
1. Introduction
Phthalazine derivatives are an important class of heterocyclic compounds that have garnered significant attention due to their diverse range of biological activities and pharmacological properties. Heterocyclic compounds are organic compounds that have structures containing ring systems made up of carbon atoms and at least one or more heteroatoms (Joule & Mills, Citation2000). They have been proven to possess a diverse array of pharmaceutical and medicinal uses. Moreover, they can be found in natural products from plants and animals such as chlorophyll, folic acid, haemoglobin, amino acids, nucleic acids and vitamins (Eicher, Hauptmann, & Speicher, Citation2012; Teran, Besada, Vila, Costas-Lago, & Ter, Citation2019). In addition, phthalazines derivatives have garnered significant interest owing to their intriguing biological features, pharmacological applications, and potential for therapeutic development, particularly in the realm of antibacterial agents (Moustafa, El‐Sayed, Abd El‐Hady, Haikal, & El‐Hashash, Citation2016; Sangshetti et al., Citation2019), antifungal (Hashash, Citation2014), anti-inflammatory activities (Vila, Besada, Costas, Costas-Lago, & Terán, Citation2015), antiviral (Lu et al., Citation2018), antileishmanial (Romero, Rodríguez, Oviedo, & Lopez, Citation2019), antidepressant (Inoue et al., Citation2015), antitumor (Wasfy, Aly, Behalo, & Mohamed, Citation2020), anticancer (Behalo, Gad El-Karim, & Rafaat, Citation2017; Khalifa et al., Citation2021), and vascular endothelial growth factor receptor 2 (VEGFR-2) activities (El Adl, Ibrahim, Khedr, Abulkhair, & Eissa, Citation2022; El Rayes, El-Enany, Ali, Ibrahim, & Nafie, Citation2022). More specifically, the anticancer activity of phthalazine derivatives has been described in various research. Hybrid molecules of steroids and phthalazine were shown to exhibit antiproliferative activity in a dose-dependent manner on various cancer cell lines (Tantawy et al., Citation2023). New phthalazine derivatives analogues of thalidomide showed far better antiproliferative activity compared to thalidomide against HepG-2, MCF7, and PC3 cell lines where the derivatives were shown to decrease the expression of caspase-8, vascular endothelial growth factor (VEGF), NF-κB P65, and TNF-α in HepG-2 cells (Kotb et al., Citation2022). Phthalazinone-dithiocarbamate hybrids were described in another work and proved significant antiproliferative activity against A2780, NCI-H460, and MCF7 cell lines (Vila, Besada, Brea, Loza, & Terán, Citation2022). Additionally, phthalazino[1,2-b]-quinazolinone derivatives were proven to possess broad spectrum histone deacetylase (HDAC) inhibition in the nanomolar range with subsequent activity against solid tumours (Liu, Zhang, Wang, Wang, & Gou, Citation2022). More importantly, the phthalazine derivative BMN-673 was found to increase cell death through inhibition of DNA repair essentially through trapping of PARP-1 and PARP-1 catalytic inhibition in breast cancer cells (Sethy & Kundu, Citation2022). Microwave-assisted synthesis is a contemporary organic synthesis technique that offers several advantages for heterocyclic compounds. These advantages include an accelerated reaction rate, reduced reaction time, and increased efficiency in synthesis methods. Additionally, the ability to control reaction parameters, such as temperature, can lead to enhanced yields and higher purity yields. Furthermore, the use of microwave-assisted synthesis allows for milder reaction conditions. Lastly, greener synthesis methods typically require lower energy consumption compared to conventional methods (Daştan, Kulkarni, & Török, Citation2012; Majumder, Gupta, & Jain, Citation2013). Previous studies have documented many aspects of organic processes, including the production of rings by cyclization as well as cycloaddition and nucleophilic substitution reactions. Multiple studies have documented the successful synthesis of phthalazine derivatives using microwave irradiation, resulting in high yields (Cao, Qu, Burton, & Rivero, Citation2008; Rao, Basak, Deb, Sharma, & Reddy, Citation2013). The field of organic synthesis has garnered considerable interest in recent times due to its application in the production of a wide range of organic compounds, including heterocyclic compounds, including compounds that are commonly found in natural products and possess a wide array of pharmacological and medicinal properties, making them valuable for synthetic purposes (Al Mousawi, El-Apasery, & Elnagdi, Citation2008; Al Saleh, Hilmy, El-Apasery, & Elnagdi, Citation2006). The objective of this study is to demonstrate the synthesis and reactions of chlorophthalazine derivatives using various reagents, including hydrazine derivatives, primary and secondary amines, and active methylene compounds. The aim is to generate novel phthalazine derivatives through the utilization of green protocol microwave irradiation methods and, subsequently, compare the yield and reaction time of the resulting products obtained from conventional methods with those obtained through microwave irradiation methods. Next, the anticancer and selective activity of compounds and doxorubicin against three cell lines, and normal fibroblasts are tested. Furthermore, a docking study is also performed to get insight into their mode of action as potential inhibitors of PARP-1 and identify the molecular mechanism of their anticancer activity.
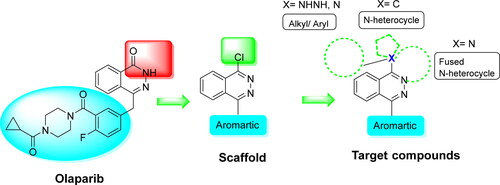
Poly(ADP-ribose) polymerase (PARP) is an enzyme involved in DNA repair. PARP inhibitors have shown promising results in both clinical trials and practice for the treatment of ovarian, breast, prostate, and pancreatic cancers (Rose, Burgess, O'Byrne, Richard, & Bolderson, Citation2020). In continuation of our work that showed the promising anticancer activity of a series of phthalazinone and phthalazine derivatives and emphasized the importance of the phthalazinone scaffold in binding the PARP-1 target and the potential of exploring the substitution in position 1, a series of 1-substituted-amino and 1-N-heterocycle phthalazinone were designed and tested (Abubshait et al., Citation2022). Our designed compounds are analogs of the anticancer drug Olaparib and would benefit from the simultaneous 1,4 substitution and the amino functionality in the 4-position, which may result in increased target selectivity and decreased toxicity ().
Figure 1. Compound design based on structure similarity with the anticancer drug, olaparib.
2. Materials and methods
2.1. General condition
All the solvents and reagents used were purchased from Sigma-Aldrich Chemicals, USA. The microwave-assisted method is carried out by using a CEM microwave (MARS™ 6 Synthesis) parallel and Scale-up microwave synthesizer by CEM company in the USA, at 300–400 W and 70–78.37 °C. Melting points determined via the Stuart SMP30 Digital Advanced MP apparatus are uncorrected. Fourier transform infrared spectroscopy (FT-IR) spectra were recorded on an IR Affinity spectrophotometer from Shimadzu. The sample prepared on glass plate contains solid KBr. Nuclear magnetic resonance (NMR) spectra were recorded by a BRUKER spectrometer (400MHZ) by using dimethylsulphoxide (DMSO‐d6) as solvent, chemical shifts (δ) in part per million (ppm) using tetramethylsilane (TMS) as internal standard. The mass spectra were carried out on LC/MS spectroscopy (LCMS-8040 Shimadzu, model CAT-30A). Elemental microanalysis was done on the Carlo Erba analyser model 110.
2.2. Synthetic chemistry procedure
2.2.1. Synthesis of 1-chloro-4-(4-isopropyl-phenyl)-phthalazine (2)
Compound 1 [4-(4-isopropyl-phenyl)-2H-phthalazin-1-one] was confirmed before, as in the previous paper (Abubshait et al., Citation2022). A mixture of 1 (2.64 gm, 10 mmol) dissolved in (30 ml) of dry benzene, phosphorus oxychloride (3.00 ml, 30 mmol), and phosphorus pentachloride (2.08 gm,10 mmol), under the air of nitrogen gas, was refluxed (30 min). Then the mixture poured in crushed ice with NaHCO3 to give compound 2 after filtered and crystallized from dry benzene to give beige powder; mp.: 240–242 °C; FT‐IR (KBr, ν, cm−1): 2922, 2865 (-CH, -CH3), 3068-3010 (=CH, Ar), 1635, 1448 (C = C); 1H NMR, δ (ppm) in DMSO-d6: 1.28 (d, 6H, 2CH3), 2.98 (m, 1H, -CH(CH3)2), 7.24-8.54 (m, 8H, -CH-, ArH); 13C NMR, δ (ppm) in DMSO-d6: 23.91 (2 C, -CH3), 30.9 (1 C, -CH(CH3)2), 126.7, 127.5, 128.5, 132.0, 148.0 (10 C, =CH-, Ar), 130.6, 134.0, 151.6 (3 C,=CH-, pyridazine), 171 (1 C, -C-Cl); MS (m/z): 282.09 (M+,100.0), 283.10 (M+ + 1,19.1); Anal. calcd. for C17H15ClN2/(282.77): C, 72.21; H, 5.35; N, 9.91; Cl, 12.54%; found: C, 72.22; H, 5.36; N, 9.92; Cl, 12.53%.
2.2.2. Synthesis of hydrazine derivatives (3–10)
A mixture of 2 (2.84 gm, 10 mmol) and hydrazine hydrate (0.65 ml, 20 mmol), and/or (10 mmol) phenylhydrazine (0.98 ml), 3,4-dimethylphenylhydrazine hydrochloride (1.72 gm), 3-nitrophenylhydrazine hydrochloride (1.89 gm), 2,4-dinirtophenyl hydrazine (1.98 gm), and 2-thiophene carboxylic hydrazide, benzoyl hydrazine (1.36 gm), and semicarbazide hydrochloride (1.12 gm) in (20 ml) dry benzene with Et3N (1 ml) was refluxed at 60 °C for 5–6 h (MW, 9–25 min). After that the precipitated products were collected, washed with cold benzene, dried, and then recrystallized from benzene.
2.2.2.1. [4-(4-Isopropyl-phenyl)-phthalazin-1-yl]-hydrazine (3)
White powder; mp.: 260–263 °C; FT‐IR (KBr, ν, cm−1): 2956, 2868 (-CH, -CH3), 3064-3010 (=CH, Ar), 1606, 1506 (C = C), 3304-3273 (-NH-NH2); 1H NMR, δ (ppm) in DMSO-d6: 1.24 (d, 6H, 2CH3), 3.47 (m, 1H, -CH(CH3)2), 7.34–8.75 (m, 8H, -CH-, Ar-H), 12.03, 13.01 (br, 3H, -NH-NH2); 13C NMR, δ (ppm) in DMSO-d6: 24.27 (2 C, -CH3), 33.7 (1 C, -CH(CH3)2), 125.6, 127.5, 128.7, 132.0, 143.0 (10 C, =CH-, Ar), 130.8, 135.0, 155.6 (3 C,=C-, pyridazine), 168.0 (1 C,=C-NH-NH2); MS (m/z): 278.15 (M+,100.0), 279.16 (M+ + 1, 56.0); Anal. calcd. for C17H18N4/(278.35); C, 73.35; H, 6.52; N, 20.13%; found: C, 73.33; H, 6.53; N, 20.14%.
2.2.2.2. N-[4-(4-Isopropyl-phenyl)-phthalazin-1-yl]-N'-phenyl-hydrazine (4)
Brown powder; mp.:147–149 °C; FT‐IR (KBr, ν, cm−1): 2957, 2862 (-CH, -CH3), 3050-3019 (=CH, Ar), 1606, 1491 (C = C), 3320-3210 (-NH-NH); 1H NMR, δ (ppm) in DMSO-d6: 2.48 (d, 6H, 2CH3), 3.42 (m, 1H, -CH(CH3)2), 6.71–8.55 (m, 13H, -CH-, ArH), 11.86 (br, 2H, -NH-NH); 13C NMR, δ (ppm) in DMSO-d6: 24.4 (2 C, -CH3), 33.7 (1 C, -CH(CH3)2), 126.4, 128.4, 128.7, 132.0 (15 C, =CH-, Ar), 130.0, 134.0, 155.6 (3 C,=C-, pyridazine), 147.3, 166.0 (2 C, =C-NH-NH-C=); MS (m/z): 354.18 (M+,100.0), 355.19 (M+ + 1, 25.1); Anal. calcd. for C23H22N4/(354.45): C, 77.94; H, 6.26; N, 15.81%; found: C, 78.00; H, 6.27; N, 15.83%.
2.2.2.3. N-[4-(4-Isopropyl-phenyl)-phthalazin-1-yl]-N'-(3,4-dimethyl phenyl-hydrazine (5)
Light brown powder; mp.:115-116 °C; FT‐IR(KBr, ν, cm−1): 2956, 2868 (-CH, -CH3), 3060-3022 (=CH, Ar), 1606, 1475 (C = C), 3320-3208 (-NH-NH); 1H NMR, δ (ppm) in DMSO-d6: 2.09 (d, 6H, 2CH3), 2.50 (s, 6H, 2CH3), 3.43 (m, 1H, -CH(CH3)2), 7.2–8.00 (m, 11H, -CH-, ArH), 8.40 (br, 2H, -NH-NH-); 13C NMR, δ (ppm) in DMSO-d6: 8.9 (2 C, -CH3), 20.0 (2 C, -CH3), 39.5 (1 C, -CH(CH3)2), 123.9, 127.8, 128.7, 130.4 (15 C, =CH-, Ar), 130.0, 135.0, 155.6 (3 C,=CH-, pyridazine), 167.1, 145.2 (2 C, =C-NH-NH-C=); MS (m/z): 382.22 (M+,100.0), 381.22 (M+ + 1,47.7); Anal. calcd. for C25H26N4/(382.50); C, 78.50; H, 6.85; N, 14.65%; found: C, 78.48; H, 6.85; N, 14.67%.
2.2.2.4. N-[4-(4-Isopropyl-phenyl)-phthalazin-1-yl]-N'-(3-nitro-phenyl)-hydrazine (6)
Light orange powder; mp.: 196–198 °C; FT‐IR (KBr, ν, cm−1): 2933, 2850 (-CH, -CH3), 3088-3010 (=CH, Ar), 1599, 1457 (C = C), 1550,1372 (-NO2), 3321-3319 (-NH-NH); 1H NMR, δ (ppm) in DMSO-d6: 1.26 (d, 6H, 2CH3), 3.42 (m, 1H, -CH(CH3)2), 7.35-8.91 (m, 12H, -CH-, ArH), 10.33,12.80 (br, 2H, -NH-NH-); 13C NMR, δ (ppm) in DMSO-d6: 24.3 (2 C, -CH3), 32.7 (1 C, -CH(CH3)2), 108.4, 116, 121, 128.8, 132.0 (14 C, =CH-, Ar), 130.0, 133.0, 155.6 (3 C,=CH-, pyridazine), 148.9 (1 C,=CH-NO2), 147.5, 169.0 (2 C, =C-NH-NH-C=); MS (m/z): 399.17 (M+,62.4), 401.17 (M+ + 2,16.1), 401.18 (M+2,3.2); Anal. calcd. for C23H21N5O2/(399.45): C, 69.16; H, 5.30; N, 17.53%; found: C, 69.16; H, 5.28; N, 17.52%.
2.2.2.5. N-[4-(4-Isopropyl-phenyl)-phthalazin-1-yl]-N'-(2,4-dinitro-phenyl)-hydrazine (7)
Brown powder; mp.: 186–188 °C; FT‐IR (KBr, ν, cm−1): 2930, 2853 (-CH, -CH3), 3078-3010 (=CH, Ar), 1597, 1458 (C = C), 1550,1370 (-NO2), 3421-3319 (-NH-NH); 1H NMR, δ (ppm) in DMSO-d6: 1.29 (d, 6H, 2CH3), 3.12 (m, 1H, -CH(CH3)2), 7.19–8.90 (m, 11H, -CH-, ArH), 10.33,12.81 (br, 2H, -NH-NH-); 13C NMR, δ (ppm) in DMSO-d6: 24.4 (2 C, -CH3), 31.7 (1 C, -CH(CH3)2), 113.4, 117, 126.7, 127, 133.7,148.7 (15 C, =CH-, Ar), 143.6 (1 C,=CH-, pyridazine), 139.9 (2 C,=CH-NO2), 143.5, 166.0 (2 C, =C-NH-NH-C=); MS (m/z): 444.15 (M+,62.4), 446.17 (M+ + 2,16.1), Anal. calcd. for C23H20N6O4/(444.44): C, 62.16; H, 4.54; N, 18.91%; found: C, 62.19; H, 5.58; N, 18.82%.
2.2.2.6. N-[4-(4-isopropyl-phenyl)-phthalazin-1-yl]-hydrazide-N'-thiophene-2-carboxylic acid (8)
Light brown powder; mp.: 190–192 °C; FT‐IR (KBr, ν, cm−1): 2930, 2852 (-CH, -CH3), 3079-3010 (=CH, Ar), 1599, 1457 (C = C), 3421-3319 (-NH-NH); 1680 (C = O); 1H NMR, δ (ppm) in DMSO-d6: 1.26 (d, 6H, 2CH3), 3.42 (m, 1H, -CH(CH3)2), 7.06–7.93 (m, 11H, -CH-, ArH), 10.33,12.80 (br, 2H, -NH-NH-); 13C NMR, δ (ppm) in DMSO-d6: 24.4 (2 C, -CH3), 31.7 (1 C, -CH(CH3)2), 119, 126, 128.5, 133.0, 136.7 (13 C, =CH-, Ar), 117.0, 127.0,143.3 (3 C,=CH-, pyridazine),145.5 (1C, -C-CO) 167.3 (1 C,-C = O), 166.0 (1 C, NH-NH-C=); MS (m/z): 388.49 (M+,100), 389.14.17 (M+ + 1,15.1), Anal. calcd. For C22H20N4OS/(388.49): C, 68.02; H, 5.19; N, 14.42; S, 8.25%; found: C, 68.05; H, 5.20; N, 14.52, S, 8.22%.
2.2.2.7. 6-(4-Isopropyl-phenyl)-3-phenyl-[1,2,4]triazolo[3,4-a]-phthalazine (9)
Light orange powder; mp.: 186–188 °C; FT‐IR (KBr, ν, cm−1): 2928, 2850 (-CH, -CH3), 3086-3011 (=CH, Ar), 1597, 1459 (C = C); 1H NMR, δ (ppm) in DMSO-d6: 1.29 (d, 6H, 2CH3), 3.12 (m, 1H, -CH(CH3)2), 7.19–7.93 (m, 13H, -CH-, ArH); 13C NMR, δ (ppm) in DMSO-d6: 24.2 (2 C, -CH3), 31.7 (1 C, -CH(CH3)2), 126.1, 128.8, 129.0, 133.0, 136.5 (18 C,=CH-,Ar, pyridazine), 127.2 (2 C,=CH-, pyridazine), 148.1 (1 C,=CH-, triazole), 152.1 (2 C, =C-N-N-C=); MS (m/z): 364.44 (M+,100), 365.40 (M+ + 1,18.1), Anal. calcd. for C24H20N4/(364.44): C, 79.10; H, 5.53; N, 15.37%; found: C, 79.14; H, 5.55; N, 15.32%.
2.2.2.8. 6-(4-Isopropyl-phenyl)-[1,2,4]triazolo[3,4-a]-phthalazin-3-yl-amine (10)
Light yellow; mp.: 176–178 °C; FT‐IR (KBr, ν, cm−1): 2932, 2853 (-CH, -CH3), 3090-3030 (=CH, Ar), 1595, 1457 (C = C), 3386-3330 (-NH2); 1H NMR, δ (ppm) in DMSO-d6: 1.29 (d, 6H, 2CH3), 3.12 (m, 1H, -CH(CH3)2), 7.19-7.93 (m, 8H, -CH-, ArH), 10.3 (br, 2H, -NH2); 13C NMR, δ (ppm) in DMSO-d6: 24.3 (2 C, -CH3), 31.7 (1 C, -CH(CH3)2), 126.7,133.0,148.7 (10 C, =CH-, Ar), 127.0, 125.1 (4 C,=CH-, pyridazine), 148.9 (1 C,=C-NH2); MS (m/z): 303.36 (M+,65.4), 305.16 (M+ + 2,16.1), Anal. calcd. for C18H17N5/(303.36): C, 71.27; H, 5.65; N, 23.09%; found: C, 71.29; H, 5.69; N, 23.10%.
2.2.3. Synthesis of compound (11–17)
A mixture of (2.84 gm, 10 mmol) of 2 and (10 mmol) of methyl-amine (0.31 ml), aniline (0.93 ml), p-toluidine (1.07 gm), p-anisidine (1.23 gm), β-naphthylamine (1.43 gm), anthranilic acid (1.37 gm) and diphenylamine (1.69 gm) in dry benzene (30 ml) placed in an oil bath and refluxed 4–6 h at 80 °C and (MW, 9–20 min). The residues were filtered to crystalline from, the mixture was extracted with ether, and ether layer was dried with anhydrous MgSO4. Finally, the residue was distilled off to be crystallized from a suitable solvent to give 11–17.
2.2.3.1. [4-(4-Isopropyl-phenyl)-phthalazin-1-yl]-methyl-amine (11)
Beige powder; mp.:160–162 °C; FT‐IR (KBr, ν, cm−1): 2970, 2868 (-CH, -CH3), 3070-3010 (=CH, Ar), 1607, 1456 (C = C), 3450 (-NH), 1188 (-C-N); 1H NMR, δ (ppm) in DMSO-d6: 1.27 (d, 6H, 2CH3), 2.31 (d, 3H, -CH3), 3.16 (m, 1H, -CH(CH3)2), 7.19–8.03 (m, 8H, -CH-, ArH), 10.57 (br, 1H, -NH-); 13C NMR, δ (ppm) in DMSO-d6: 24.1 (2 C, -CH3), 35.3 (1 C, -CH3), 31.5 (1 C, -CH(CH3)2), 126.7, 127.4, 132.1,133.4 (10 C, =CH-, Ar),117, 127.1, 143.1 (3 C,=CH-, pyridazine), 166.0 (1 C, =C-NH-); MS (m/z): 277.36 (M+,98.0); Anal. calcd. for C18H19N3/(277.36): C, 77.95; H, 6.90; N, 15.15%; found: C, 77.93; H, 6.87; N, 15.17%.
2.2.3.2. [4-(4-Isopropyl-phenyl)-phthalazin-1-yl]-phenyl-amine (12)
Beige powder; mp.:150–151 °C; FT‐IR (KBr, ν, cm−1): 2955, 2870 (-CH, -CH3), 3061-3010 (=CH, Ar), 1607, 1456 (C = C), 3450 (-NH), 1188 (-C-N); 1H NMR, δ (ppm) in DMSO-d6: 1.27 (d, 6H, 2CH3), 3.46 (m, 1H, -CH(CH3)2), 6.58–8.33 (m, 13H, -CH-, ArH), 11.57 (br, 1H, -NH-); 13C NMR, δ (ppm) in DMSO-d6: 24.3 (2 C, -CH3), 31.7 (1 C, -CH(CH3)2), 115.4, 117.0, 126.9, 128.4, 129.4, 133.1, 147.1 (15 C, =CH-, Ar), 131.0, 133.3, 151.1 (3 C,=CH-, pyridazine), 166.2, 166.0 (2 C, =C-NH-C=); MS (m/z): 339.17 (M+,61.0); Anal. calcd. for C23H21N3/(339.43): C, 81.38; H, 6.24; N, 12.38%; found: C, 81.40; H, 6.22; N, 12.40%.
2.2.3.3. [4-(4-Isopropyl-phenyl)-phthalazin-1-yl]-p-tolyl-amine (13)
Beige powder; mp.:140–141 °C; FT‐IR (KBr, ν, cm−1): 2965, 2868 (-CH, -CH3), 3050-3020 (=CH, Ar), 1604, 1479 (C = C), 3450 (-NH), 1170 (-C-N); 1H NMR, δ (ppm) in DMSO-d6: 2.18 (d, 6H, 2CH3), 2.51 (s,3H, 2CH3), 3.54 (m, 1H, -CH(CH3)2), 6.71–8.07 (m, 12H, -CH-, ArH), 9.0 (br, 1H, -NH-); 13C NMR, δ (ppm) in DMSO-d6: 20.7 (1 C, -CH3), 24.3 (2 C, -CH3), 34.7 (1 C, -CH(CH3)2), 117.0, 128.4, 129.4, 133.1, 141.1 (15 C, =CH-, Ar), 130.0, 133.3, 151.1 (3 C,=CH-, pyridazine), 168.2, 169.0 (2 C,=C-NH-C=); MS (m/z): 353.19 (M+,100.0), 350.19 (M+-3,7.8); Anal. calcd. for C24H23N3/(353.46): C, 81.55; H, 6.56; N, 11.89%; found: C, 81.54; H, 6.56; N, 11.90%.
2.2.3.4. [4-(4-Isopropyl-phenyl)-phthalazin-1-yl]-(4-methoxy-phenyl)-amine (14)
Light purple powder; mp.: 126–128 °C; FT‐IR (KBr, ν, cm−1): 2960, 2868 (-CH, -CH3), 3060-3020 (=CH, Ar), 1600, 1475 (C = C), 1270 (-C-O-C), 3450 (-NH), 1170 (-C-N); 1H NMR, δ (ppm) in DMSO-d6: 1.27 (d, 6H, 2CH3), 3.46 (m, 1H, -CH(CH3)2), 1.27 (s,3H, -OCH3), 6.77-8.07 (m, 12H, -CH-, ArH), 11.34 (br, 1H, -NH-); 13C NMR, δ (ppm) in DMSO-d6: 24.3 (2 C, -CH3), 31.7 (1 C, -CH(CH3)2), 55.8 (1 C, -OCH3), 115.4, 118, 125.6, 128.4, 129.4, 133.1, 147.1 (14 C, =CH-, Ar), 131.0, 133.3, 151.1 (3 C,=CH-, pyridazine), 154.0 (1 C, =C-OCH3), 139.0, 169.0 (2 C, =C-NH-C=); MS (m/z): 369.18 (M+,100.0), 370.19 (M+ + 1,27.1); Anal. calcd. for C24H23N3O/(369.46): C, 78.02; H, 6.27; N, 11.37%; found: C, 78.03; H, 6.27; N, 11.40%.
2.2.3.5. [4-(4-Isopropyl-phenyl)-phthalazin-1-yl]-naphthalen-2-yl-amine (15)
Beige powder; mp.:180–183 °C; FT‐IR (KBr, ν, cm−1): 2958, 2878 (-CH, -CH3), 3071-3030 (=CH, Ar), 1609, 1459 (C = C), 3450 (-NH), 1188 (-C-N); 1H NMR, δ (ppm) in DMSO-d6: 1.28 (d, 6H, 2CH3), 3.45 (m, 1H, -CH(CH3)2), 6.76–8.01 (m, 15H, -CH-, ArH), 10.57 (br, 1H, -NH-); 13C NMR, δ (ppm) in DMSO-d6: 24.3 (2 C, -CH3), 31.7 (1 C, -CH(CH3)2), 115.4, 119.0, 126.7, 127.4, 133.1, 148.1 (19 C, =CH-, Ar), 117.1, 127.3, 127.1 (3 C,=CH-, pyridazine), 142.6, 166.6 (2 C, =C-NH-C=); MS (m/z): 389.19 (M+,100.0), 390.19 (M+ + 1,27.1); Anal. calcd. for C27H23N3/(389.49): C, 83.26; H, 5.95; N, 10.79%; found: C, 83.21; H, 5.98; N, 10.80%.
2.2.3.6. 5-(4-Isopropyl-phenyl)-6,6a,12-triaza-benzo[a]anthracen-7-one (16)
White powder; mp.: 190–191 °C; FT‐IR (KBr, ν, cm−1): 2957, 2862 (-CH, -CH3), 3060-3010 (=CH, Ar), 1600, 1475 (C = C), 1670 (C = O); 1H NMR, δ (ppm) in DMSO-d6: 1.20 (d, 6H, 2CH3), 3.42 (m, 1H, -CH(CH3)2), 7.2–7.5 (m, 12H, -CH-, ArH); 13C NMR, δ (ppm) in DMSO-d6: 24.4 (2 C, -CH3), 31.7 (1 C, -CH(CH3)2), 110, 116, 126.4, 128.4, 131, 133 (16 C, =CH-, Ar), 134.3, 155.6 (2 C,=CH, pyridazine), 151, 169 (2 C, -C = N); 170 (1 C, -C = O); MS (m/z): 365.15 (M+,100), 366.16 (M+ + 1,27.0) Anal. calcd. for C24H19N3O(365.43): C, 78.88; H, 5.24; N, 11.50%; found: C, 78.90; H, 5.26; N, 11.52%.
2.2.3.7. [4-(4-Isopropyl-phenyl)-phthalazin-1-yl]-diphenyl-amine (17)
White powder; mp.:203–204 °C; FT‐IR(KBr, ν, cm−1): 2962, 2852 (-CH, -CH3), 3070-3010 (=CH, Ar), 1600, 1475 (C = C), 1360 (-C-N); 1H NMR, δ (ppm) in DMSO-d6: 1.25 (d, 6H, 2CH3), 3.43 (m, 1H, -CH(CH3)2), 7.35–8.07 (m, 18H, -CH-, ArH),13C NMR, δ (ppm) in DMSO-d6: 24.3 (2 C, -CH3), 33.7 (1 C, -CH(CH3)2), 117, 120, 125.6, 127, 128.8, 129.6, 133.1, 147.1 (20 C, =CH-, Ar), 131, 133.3, 151.1 (3 C,=CH-, pyridazine), 143, 169 (3 C, -N-C=); MS (m/z): 415.20 (M+,100.0), 416.21 (M+ + 1,15.1); Anal. calcd. for C29H25N3/(415.53): C, 83.82; H, 6.06; N, 10.11%; found: C, 83.84; H, 6.04; N, 10.09%.
2.2.4. Synthesis compounds pyrazol phthalazin derivatives (18–21)
A mixture (2.84 gm,10 mmol) of 2 (10 mmol) of acetyl acetone (1.00 ml), and/or ethyl acetoacetate (1.27 ml), ethyl cyanoacetate (1.13 ml), and malononitrile (0.55 gm) was dissolved in 30 ml ethanol with C2H5ONa, then refluxed in an oil bath for 5 h at 80 °C (MW, 10 min). After an excess solvent is distilled, off, poured into cold water, then extracted with diethyl ether. The solid crystallized from benzene, then dissolved in 30 ml of ethanol, and (0.9 ml, 20 mmol) of hydrazine hydrate, and refluxed to give 18–21, The separated solid crystallized from a suitable solvent.
2.2.4.1. 1-(3,5-Dimethyl-4H-pyrazol-4-(4-isopropyl-phenyl)-phthalazine (18)
White crystals; mp.:270–271 °C; FT‐IR(KBr, ν, cm−1): 2962,2852 (-CH, -CH3), 3060, 3010 (=CH, Ar), 1598,1440 (C = C), 1610 (C-N-N); 1H NMR, δ (ppm) in DMSO-d6: 1.29 (d, 6H, 2-CH3), 2.00 (s, 6H, 2-CH3, pyrazole), 2.50 (d,1H, -CH-, pyrazole), 3.12 (m, 1H, -CH(CH3)2), 7.4–8.3 (m, 8H, -CH-, ArH); 13C NMR, δ (ppm) in DMSO-d6: 18.2 (2 C, -CH3, pyrazole), 24.3 (2 C, -CH3), 33.7 (1 C, -CH(CH3)2), 39.9 (1 C, -CH-, pyrazole), 126.5, 127.1, 128.7, 129 (10 C, =CH-, Ar), 132, 134.5, 150.6 (4 C,=CH-, pyridazine), 160 (2 C, -C-CH3, pyrazole); MS (m/z): 342.18 (M+,100.0), 341.19 (M+-1,24.8); Anal. calcd. for C22H22N4/(342.44): C, 77.16; H, 6.48; N, 16.36%; found: C, 77.18; H, 6.45; N, 16.38%.
2.2.4.2. 4-[4-(4-Isopropyl-phenyl)-phthalazin-1-yl]-5-methyl-2,4-dihydro-pyrazol-3-one (19)
White crystal; mp.:250–252 °C; FT‐IR(KBr, ν, cm−1): 2963,2853 (-CH, -CH3), 3067-3020 (=CH, Ar), 1598,1440 (C = C), 1680 (C = O), 1610 (C-N-N); 1H NMR, δ (ppm) in DMSO-d6: 1.29 (d, 6H, 2CH3), 2.00 (s, 3H, -CH3), 3.43 (m, 1H, -CH(CH3)2), 5.21 (s,1H, -CH-, pyrazole), 7.243–8.3 (m, 8H, -CH-, ArH), 12.85 (br, 1H, -NH); 13C NMR, δ (ppm) in DMSO-d6: 15.3 (1 C, -CH3), 24.3 (2 C, -CH3), 33.8 (1 C, -CH(CH3)2), 40.3 (1 C, -CH-, pyrazole), 126.6, 127, 128.3, 129.7, 134 (10 C, =CH-, Ar), 132, 147, 150 (4 C,=CH-, pyridazine), 155.6 (1 C, -CH-, pyrazole), 159.7 (1 C, -C = O); MS (m/z): 344.1 6 (M+, 100.0); 345.17 (M+ + 1, 20); Anal. calcd. for C21H20N4O/(344.41): C, 73.23; H, 5.85; N, 16.27%; found: C, 73.20; H, 5.86; N, 16.27%.
2.2.4.3. 5-Amino-4-[4-(4-isopropyl-phenyl)-phthalazin-1-yl]-2,4-dihydro-pyrazol-3-one (20)
Light brown crystals; mp.:169–170 °C; FT‐IR(KBr, ν, cm−1): 2962,2850 (-CH, -CH3), 3065-3025 (=CH, Ar), 1625,1475 (C = C), 1682 (C = O), 3409, 3350 (-NH2), 1625 (C-N-N); 1H NMR, δ (ppm) in DMSO-d6: 1.28 (d, 6H, 2CH3), 3.01 (m, 1H, -CH(CH3)2), 4.7 (s,1H, -CH-, pyrazole), 7.43–8.34 (m, 8H, -CH-, ArH), 12.85, 11.9 (br, 3H, -NH2, -NH-); 13C NMR, δ (ppm) in DMSO-d6: 24.3 (2 C, -CH3), 33.8 (1 C, -CH(CH3)2), 51.0 (1 C, -CH-, pyrazole), 126.5, 127, 128.3, 128.7, 129 (10 C, =CH-, Ar); 132.6, 134.5, 155.6 (4 C,=CH-, pyridazine),149.7 (1C, =C-NH2, pyrazole), 159.7 (1 C, -C = O); MS (m/z): 345.16 (M+,51.39), 347.17 (M+ + 2,31.0); Anal. calcd. for C20H19N5O/(345.40):C, 69.55; H, 5.54; N, 20.28%; found: C, 69.57; H, 5.55; N, 20.29%.
2.2.4.4. 4-[4-(4-Isopropyl-phenyl)-phthalazin-1-yl]-4H-pyrazole-3,5-diamine (21)
Light brown powder; mp.:172–173 °C; FT‐IR(KBr, ν, cm−1): 2960,2856 (-CH, -CH3), 3070-3030 (=CH, Ar), 1600,1465 (C = C), 3450,3300 (-NH2), 1625 (C-N-N); 1H NMR, δ (ppm) in DMSO-d6: 1.28 (d, 6H, 2CH3), 3.42 (m, 1H, -CH(CH3)2), 2.65 (d,1H, -CH-, pyrazole), 7.36–8.27 (m, 8H, -CH-, ArH), 12.85, 11.87 (br, 4H, -NH2); 13C NMR, δ (ppm) in DMSO-d6: 24.4 (2 C, -CH3), 33.8 (1 C, -CH(CH3)2), 40.3 (1 C, -CH-, pyrazole), 126.5, 127.1, 128.4, 129, 146 (10 C, =CH-, Ar), 132.1, 134.1, 150 (4 C,=CH-, pyridazine), 160 (2 C,=CH-NH2); MS (m/z): 344.17 (M+,100.0), 345.18 (M+ + 1, 22.5); Anal. calcd. for C20H20N6 (344.41): C, 69.75; H, 5.85; N, 24.40%; found: C, 69.77; H, 5.88; N, 24.38%.
2.3. Biological evaluation
2.3.1. Cell culture
In the present study, we have utilized three cancer cell lines with the utmost range of 15 passages. These cell lines represent three organs starting with the breast (MCF7), colon (HCT116), and liver (HepG2). To examine the selectivity of compounds, a normal foetal lung fibroblast was used with almost 10 passages (MRC5). All cells were sources from the American Type Culture Collection (ATCC), USA. The three cancer cells were maintained in RPMI-1640 media (10% Fetal bovine serum (FBS)), while MRC5 was maintained in Eagle’s minimum essential medium (10% FBS), with the standard conditions (37 °C, 5% CO2, and 100% relative humidity (Abdalla, Abdallah, et al., Citation2020).
2.3.2. Anticancer and selectivity studies
In order to determine the anticancer activity of an investigational compound, the 19 compounds, in addition to doxorubicin were tested using the MTT assay according to a previous study (Abdalla, Qattan, et al., Citation2020). Each cell line was subcultured to 80%–90% confluency before adding each compound or doxorubicin at final concentrations of 0–50 µM for 72h. Next, MTT was added to each well including negative controls and incubated for 3h at 37 °C. MTT was then removed, and the remaining precipitation was dissolved with DMSO. The absorbance (A550) for each well/concentration was read on multi-plate reader (Bio-Rad, PR 4100, Hercules, CA, USA), and the inhibition concentration (IC50) was calculated using GraphPad Prism software (version 5). The selectivity index (SI) for each of the 19 compounds and doxorubicin was calculated by dividing the compound IC50 against MRC-5 cells/IC50 against either MCF7, HCT116 or HepG2 cells (Abdalla et al., Citation2021).
2.4. Molecular modeling
Molecular docking simulation and ligand binding energy calculation were performed using the molecular operating environment (MOE) 2014 (CCG®; Montreal, Canada). PyMol 2.0.6 Graphical Software (Schrödinger®, New York, NY, USA) was used for binding interaction visualization and figures generation. Ligand preparation, energy minimization, and potential energy calculation were performed using MOE interface. The crystal structure of human PARP-1 (PDB code: 7aad), co-crystallized with Olaparib was selected for the docking studies. The selected target and ligands were prepared using protein preparation and LigX tools, respectively. The protein underwent three-dimensional protonation, auto-correction of atom types and connections, and partial charge assignment at a physiological neutral pH of 7.4. The ligand PDB file was modified by adding hydrogens, the partial charges and ionization state were calculated, and was energy minimized using MMFF94X forcefield. The active site for docking was defined by residues within 5 Angstrom from the co-crystallized ligand. The docking procedure was executed using the MOE dock tool within the MOE software, utilizing the default parameters. This process utilized rigid docking, in which the protein remained inflexible while the ligands were kept flexible and the ligand’s conformation was generated via ligand placement and bond rotation. The docking output was set to 10 docking poses per ligand. The results were assessed by identifying potential ligand interactions with the protein active site using the MOE–ligand interactions wizard and bond distance calculation in PyMol. The binding score (S score) in MOE and the comparative docking with the crystal ligand were also used to identify the best binders. The selection was made using a comprehensive approach that included the best docking score, low root mean square deviation (RMSDs) compared to the co-crystalline ligand, and important interactions with the active site.
3. Results and discussion
3.1. Chemistry
Phthalazine and phthalazinone derivatives have garnered significant attention in medicinal chemistry and drug discovery and had been reported to have a wide range of biological actions and medical applications such as antimicrobial (Ibrahim, Eldehna, Abdel-Aziz, Elaasser, & Abdel-Aziz, Citation2014; Mourad, Makhlouf, Soliman, & Mohamed, Citation2020), anticancer (El Sayed et al., Citation2024; Emam, Rayes, Ali, Soliman, & Nafie, Citation2023; Vila et al., Citation2022), antitumor (Eldehna, Ibrahim, Abdel-Aziz, Farrag, & Youssef, Citation2015; Marzouk, Shaker, Hafiz, & El-Baghdady, Citation2016), anti-Alzheimer’s inhibitors (Ezzat, Abdelhamid, Fouad, Abdel‐Aziz, & Allam, Citation2023), anti of acquired immunodeficiency syndrome (anti-AIDS) (Sroczyński, Malinowski, Szcześniak, & Pakulska, Citation2016), anti-inflammatory, antiproliferative agents (Hameed et al., Citation2016), anticonvulsant (Ayyad, Sakr, & El-Gamal, Citation2016), and antidiabetic (Agrawal et al., Citation2013). In addition, phthalazinone 1 was synthesized in a prior study conducted by Abubshait et al. (Citation2022). Chlorophthalazine 2 (Scheme 1) was produced by refluxing compound 1 with a combination of phosphorus pentachloride and phosphorus oxychloride (Abubshait, Citation2007; Zhang et al., Citation2010).
Scheme 1. General methods for preparation chlorophthalazine 2.
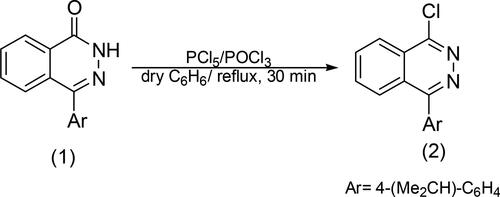
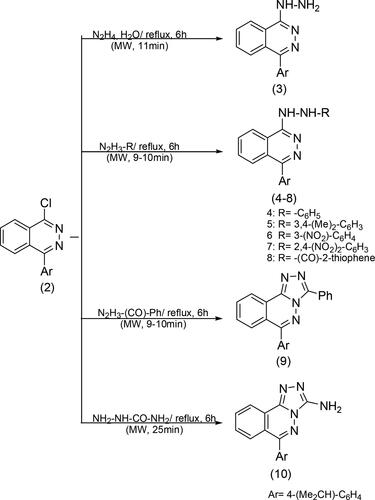
Additionally, chlorophthalazine structure for FT-IR spectra showed conforming stretching bands for aliphatic and aromatic groups, major bands at 2922, 2865 cm−1 (-CH, -CH3), 3068-3010 cm−1 (=CH, Ar), 1635, 1448 cm−1 (C = C), and disappearance of the carbonyl group of phthalazinone 1. Moreover, the (1H,13C) NMR spectra of 2 showed the presence of obvious bands for -CH3, -CH(CH3)2, related to pyridazine, aromatic groups also new protons and carbon bands are presented, at 1.28 (d, 6H, 2CH3), 2.98 (m, 1H, -CH(CH3)2), 7.24–8.54 (m, 8H, -CH-, ArH); 13C NMR, δ (ppm) 23.91 (2 C, -CH3), 30.9 (1 C, -CH(CH3)2), 126.7, 127.5, 128.5, 132.0, 148.0 (10 C, =CH-, Ar), 130.6, 134.0, 151.6 (3 C,=CH-, pyridazine), 171 (1 C, -C-Cl). The one-pot reaction of chlorophthalazine is an important synthetic application to produce a heterocyclic nucleus possessing broad pharmaceutical and biological activity (Ahmad, Varshney, Rauf, Husain, & Ahmad, Citation2014; Khidre, Mostafa, Mukhrish, Salem, & Behalo, Citation2022; Romero et al., Citation2020). This study investigates the nucleophilic displacement reactions of chlorophthalazine with different hydrazine derivatives, primary and secondary amines, and active methylene compounds in order to produce novel phthalazine derivatives. The reactions were conducted using both traditional methods and microwave irradiation to synthesize new phthalazine derivatives from 2 with high yields and a rapid reaction rate. Microwave irradiation was employed as a green and efficient technology to assist in the synthesis process (Almahli et al., Citation2018; Boraei et al., Citation2019; Bunce, Harrison, & Nammalwar, Citation2012). The use of microwave irradiation in synthetic of heterocycle compounds has several advantages over traditional synthetic methods is a promising method for accelerating reaction rates and improving yields and improve the selectivity of various organic transformations (Damera, Pagadala, Rana, & Jonnalagadda, Citation2023). The traditional synthetic methods of heterocycles have relied on multi-step processes involving distinct reaction steps and purification procedures, these conventional methods often encounter drawbacks such as decreased productivity, extended reaction times, and the production of by-products. However, using microwave irradiation as green strategy for the improved synthesis of biologically and pharmaceutically interesting annulated heterocycles has further highlighted the potential of this approach in accessing novel heterocyclic systems (Javahershenas, Makarem, & Klika, Citation2024). Scheme 2 shows the reaction of chlorophthalazine 2 with hydrazine hydrate, phenylhydrazine, 3,4-dimethylphenylhydrazine, 3-nitrophenylhydrazine, 2,4-dinitrophenylhydrazine, 2-thiophenecarboxylic hydrazine, benzoylhydrazine and semicarbazide experimented under reflux for 5–6 h and under microwave for 9–25 min in dry benzene to give 3–10, respectively. The FT-IR spectra of hydrazine derivatives showed new (-NHNH2, -NHNH-) stretching bands at regions 3421-3304 and 3319-3210 cm−1.
Scheme 2. Reactions of chlorophthalazine 2 with various hydrazine derivatives.
The displacement of chloride from chlorophthalazine can be achieved by nucleophile attacks on electron-rich groups, specifically primary and secondary amines such as methyl-amine, aniline, p-toluidine, p-anisidine, α-naphthylamine, anthranilic acid, and diphenylamine. This can be achieved through reflux for a duration of 4–6 h or microwave (MW) for 9–10 min to give the resulting products 11–17, respectively. Scheme 3 (Abou-Seri, Eldehna, Ali, & Abou El Ella, Citation2016; Abubshait, Kassab, Al-Shehri, & Abubshait, Citation2011; Behalo et al., Citation2017).
Scheme 3. Reactions of chlorophthalazine 2 with primary and secondary amines.
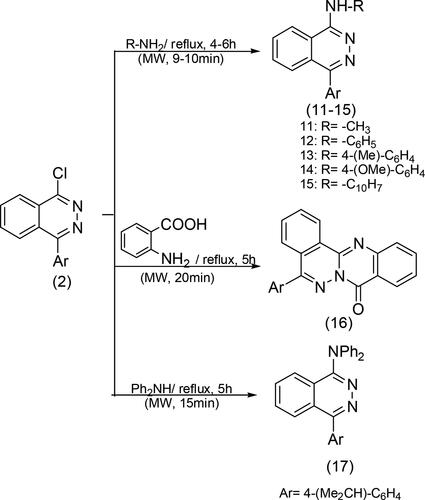
Compounds 11–17 exhibited bands at 3450 and 1188–1360 cm−1, which can be attributed to the presence of secondary and tertiary amine groups (-NH, -C-N). The reaction between compound 2 and carbon nucleophiles, in which active methylene was involved, resulted in the formation of pyrazolo derivatives. Phthalazine derivatives were obtained through the reactions of compound 2 with acetyl acetone, ethyl acetoacetate, ethyl cyanoacetate, and malononitrile in the presence of sodium ethoxide. These derivatives were further subjected to a cyclization process with hydrazine hydrate in ethanol, resulting in the formation of pyrazolo derivatives 18–21, respectively. Scheme 4 (Abubshait, Citation2007; El Shamy et al., Citation2023).
Scheme 4. General methods for preparation of compounds 18–21.
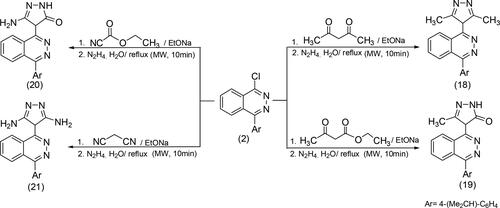
Coupling of 2 with various active methylene compounds afforded the corresponding coupling products by applying conventional methods in ethanol under reflux for 6h (Henary et al., Citation2020; Richard et al., Citation1986). In comparison, the reaction was successfully performed within a time frame under microwave irradiation. In addition to a short-time reaction, the collected products were obtained in high yields compared with the conventional method. Scheme 4 represents the pathway for preparing the target compounds through the nucleophilic attack of 2 with active methylene compounds by using MW irradiation. The synthesized compounds were found to undergo a cyclization process as a result of the nucleophilic attack within the nucleophile hydrazinehydrate which led to the formation of the corresponding pyrazolyl derivatives. A comprehensive analysis of the unique products is conducted using their spectral data. The IR spectra exhibited absorption bands within the range of 1680-1680 cm−1, which can be attributed to the (C = O) functional group associated with compounds 19 and 20. Compounds 20 and 21 exhibited new stretching bands at positions 3409-3450-3350-3000, which can be attributed to the (NH2) group. The 1H NMR spectra exhibited prominent bands corresponding to isopropyl, aliphatic, and aromatic protons. Additionally, novel bands were seen for methyl and pyrazole protons at a wavenumber of 2.00 and 2.50 ppm, respectively. The spectra of compounds 18–21 exhibit new signals for the (NH2, -NH-) group. displays the yield and reaction time (in minutes) of compounds 2–21, obtained using MW irradiation and the conventional approach.
Table 1. Yield (%) and reaction time (min) of compounds 2–21 by using MW irradiation and conventional method.
3.2. Biological evaluation
3.2.1. Anticancer and selectivity studies
To determine the anticancer effect of compounds 3–21 against MCF7, HCT116, and HepG2 cancer cells, an MTT assay was performed. The most active compound against MCF7 cells was 13 with IC50 0.286 µM, which was less effective but comparable with the IC50 of doxorubicin against MCF7 (0.041 µM), while the least active compound was No. 4 with IC50 5.370 µM. The range of anticancer activity against HCT116 was wider, as compound 6 showed IC50 0.384 µM, which was 10 times better than the anticancer effect of doxorubicin against HCT116 (IC50 3.964 µM), while compound 3 was less active against HCT116 with IC50 25.228 µM. Regarding HepG2, the most active compound was 16 with IC50 0.484 µM which was comparable with the IC50 of doxorubicin against HepG2 cells (0.381 µM), while the least active compound was 4 with IC50 5.808 µM. Thus, the most active compounds were 13, 6, and 16, respectively (). also includes the anticancer effect of each compound against MRC5 normal fibroblast cells. The IC50 ranges between 0.839 µM and 25.800 µM. This data was used to calculate the SI of each compound towards MRC5 normal fibroblast cells () compared with each of the three cell lines, and the best SI was for compound 6 (), followed by compound 13, all of which was comparable with the selectivity of doxorubicin.
Figure 2. Dose response curve of compound 6 against MCF7, HCT116, HePG2 and MRC5 cells. X-axis: dose, Y-axis: absorbance.
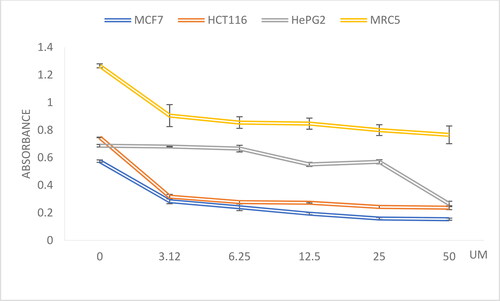
Table 2. Anticancer activity of compounds 3–21 and doxorubicin against three cell lines, and MRC5 normal fibroblast cells (MTT 72 h, IC50 “µM” ± SD, n = 3).
Table 3. Selectivity index (SI) of compounds 3–21* and doxorubicin against MRC5 normal fibroblast cells.
The best selective compounds were compound 6 selective against three cells compared to MRC5 cells, followed by compounds 13, 16, 8, and 14, best cytotoxic and selective compounds 6 and 13 ().
3.3. Molecular modeling
Molecular docking simulations performed on the synthesized compounds with poly ADP- ribose polymerase (PAPR-1) revealed a favourable binding affinity. The results confirmed the experimental IC50, which predominantly fell within the one-digit micromolar range, and successfully identified the compounds with the highest experimental activity (6, 13, 19, 20) as the best binders. Those compounds were especially perfectly aligned with the standard co-crystallized inhibitor, Olaparib in the active site ().
Figure 3. Comparative binding positions of compound 6 (green), compound 13 (blue), compound 19 (magenta), compound 20 (orange) and olaparib (red) bound to PARP-1 (7aad) active site.
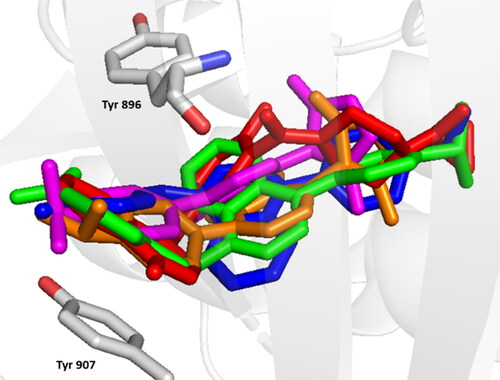
The docking studies showed that the most active compounds were able to bind the gatekeeper residues, Tyr 896, and Tyr 907, which were identified in our previous study as being crucial for enzyme binding. The initial binding with these two residues is the starting point for ligands to access the active site, and the π–π interactions formed with these residues give affinity and selectivity to the interacting ligand. In the most active compounds, one of the aromatics interacts with Tyr 907 while the phthalazinone ring performs the interaction with Tyr 896. The less lipophilic phthalazinone ring is the part of the structure that is solvent exposed while the more lipophilic isopropyl-phenyl at position-4 is directed deeper in the binding pocket which stabilizes the compounds in the active site. This is either in the tiny binding pocket of the enzyme formed by Gly 863, Ser 907 and Glu 988 in the case of compounds 19 and 20 (, respectively) or in the more spacious hydrophobic pocket formed by Tyr 710, Asp 766, Leu 769, Arg 878, Ala 880, Pro 881, and Tyr 889 in case of compounds 6 and 13 (, respectively). The substituted nitrogen moiety at position-1 on the other hand occupies the opposite side of the binding pocket. This interaction was noted for compounds 6 and 13 possibly because of the higher flexibility of the anilino or phenylhyarazino moiety compared to the more rigid pyrazole ring in compounds 19 and 20, which allows them to fit better in the tiny pocket.
Figure 4. Surface representation of compound 6 (a), compound 13 (b), compound 19 (c) and compound 20 (d) bound to PARP-1 (7aad) active site.
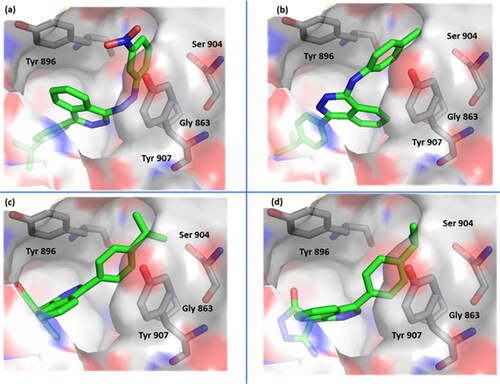
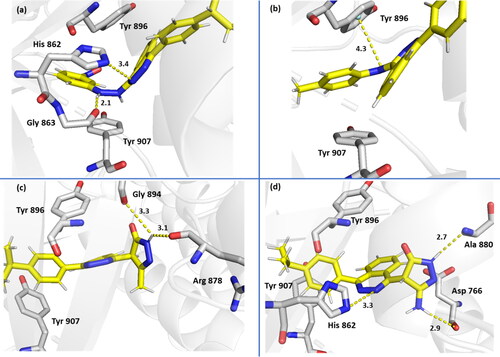
Compound 6 (IC50, 1.214) established two additional electrostatic interactions with its phthalazinone and side chain hydrazino nitrogens and His 862 sidechain and Gly 863 backbone, respectively (). Compound 13 (IC50, 2.046) did not show additional electrostatic bonds, however, a potential and characteristic π–H bonding was noted with its anilino nitrogen and Tyr 896 which may explain the comparable activity with the other compounds despite the lack of direct H-bonding (). Compound 19 (IC50, 1.803) was able to gain high affinity through H-bonding with its pyrazole ring and Arg 878 and Gly 894 backbones, where it acted as a strong H-bond donor (). Compound 20 (IC50, 1.676) was identified as one of the best binder due to formation of several H-bonds with residues in the binding pocket more specifically with its pyrazole and Ala 880 backbone and its amine and Asp 766 backbone and its phthalzinone nitrogen and His 862 sidechain ().
Figure 5. Binding interactions of compound 6 (a), compound 13 (b), compound 19 (c) and compound 20 (d) with PARP-1 (7aad) active site. Potential electrostatic interactions are represented as yellow dotted lines and are measured in angstrom.
The results of the biological testing and the docking studies formulated a primary structure–activity relationship for the phthalazine derivatives. The phthalazine core is important and is involved in anchoring the compounds at the active site. Substitution at position 1 with a phenyl ring gives more active compounds as the aromatic ring digs deeper in the active site and establishes hydrophobic interactions and π–π stacking. Substitution at position 4 with amino functionality is important for the activity, and it can accommodate aminophenyl, hydrazinophenyl, and aminohetreocycles as examples. However, increasing the steric hindrance in this part such as disubstituted amine (compound 17) or ring expansion (compound 16), reduced the activity.
The fact that PARP is overexpressed in the tested cancer cells (Siraj et al., Citation2018) together with the high affinity of the synthesized compounds, imparted a high selectivity on cancer cells compared to normal cells with compounds 6, 8, 13, 14, and 16 being the most selective. Compounds 6 and 13 especially showed selectivity comparable to the clinically used drug doxorubicin in MCF7 cells (15.17 and 41.5, respectively, compared to 60.39) and superior selectivity on HCT116 (67.89 and 2.39, respectively, compared to 0.62) and HepG2 cell lines (16.97 and 11.62, respectively, compared to 6.48). The docking scores also correlated well with the experimental activity and selectivity data. A comparison of the binding scores, average IC50, and average selectivity of the most active compounds is presented in .
Table 4. Binding scores, average IC50, and average selectivity of the most active compounds.
In conclusion, the optimization of our phthalzinone compounds through exploring the substitution on position 1 with various alkyl and aryl substituted nitrogen and nitrogen heterocycles enhanced both the affinity and selectivity to the PARP target. The docking scores were aligned with the experimental data, which validated the compounds’ target interactions and selectivity. Future research directions will focus on testing our phthalazines experimentally for PARP-1 activity together with synthesizing more derivatives that explore substitution at the other positions of the phthalazine core, which together will guide the drug design and optimization for in vivo preclinical testing.
4. Conclusions
A new series of phthalazine derivatives have been produced by utilizing environmentally friendly processes that use microwave irradiation in high yield and short reaction times. The utilization of the dynamic microwave power system resulted in highly effective heating of the materials, leading to decreased durations of chemical reactions and enhanced yields of reactions in the preparation of the target phthalazine derivatives. The synthesized phthalazine derivatives showed broad anticancer activity against three different cell lines namely, MCF7, HCT116, and HepG2. Compound 6 was identified as the most active and selective compound that showed comparable results with doxorubicin. The docking results concurred with the experimental data and highlighted the influence of substituting position 1 of the phthalazine derivatives on their binding affinity to the PARP-1 active site. Our computational analysis findings have identified our phthalazine compounds as a novel scaffold for inhibiting PARP-1.
Supplementary.pdf
Download PDF (1.1 MB)Acknowledgements
We would like to thank the College of Science and the Basic and Applied Scientific Research Center at Imam Abdulrahman Bin Faisal University for the co-workers and making the spectral analyses.
Disclosure statement
The authors declare that there is no conflict of interest regarding this publication.
References
- Abdalla, A. N., Abdallah, M. E., Aslam, A., Bader, A., Vassallo, A., De Tommasi, N., … El-Azab, A. (2020). Synergistic anti leukemia effect of a novel Hsp90 and a pan cyclin dependent kinase inhibitors. Molecules, 25(9), 2220. doi:10.3390/molecules25092220
- Abdalla, A. S., Malki, W. H., Amal Qattan, A., Shahid, I., Hossain, M. A., & Ahmed, M. (2021). Chemosensitization of HT29 and HT29-5FU cell lines by a combination of a multi-tyrosine kinase inhibitor and 5FU downregulates ABCC1 and inhibits PIK3CA in light of their importance in Saudi colorectal cancer. Molecules, 26(2), 334. doi:10.3390/molecules26020334
- Abdalla, A. N., Qattan, A., Malki, W. H., Shahid, I., Hossain, M. A., & Ahmed, M. (2020). Significance of targeting VEGFR-2 and Cyclin D1 in Luminal-A breast cancer. Molecules, 25(20), 4606. doi:10.3390/molecules25204606
- Abou-Seri, S. M., Eldehna, W. M., Ali, M. M., & Abou El Ella, D. A. (2016). 1-Piperazinylphthalazines as potential VEGFR-2 inhibitors and anticancer agents: Synthesis and in vitro biological evaluation. European Journal of Medicinal Chemistry, 107, 165–179. doi:10.1016/j.ejmech.2015.10.053
- Abubshait, S. A. (2007). An efficient synthesis and reactions of novel indolyl-pyridazinone derivatives with expected biological activity. Molecules, 12(1), 25–42. doi:10.3390/12010025
- Abubshait, S. A., Abubshait, H. A., Almalih, R., Gomaa, M. S., Nawaz, M., Ababutain, I. M., & Alghamdi, A. I. (2022). Microwave-assisted synthesis of phthalazinone derivatives with biological activity and in silico antiproliferative studies. ChemistrySelect, 7(46), e202203925. doi:10.1002/slct.202203925
- Abubshait, S. A., Kassab, R. R., Al-Shehri, A. H., & Abubshait, H. A. (2011). Synthesis and reactions of some novel 4-biphenyl-4-(2H)-phthalazin-1-one derivatives with an expected antimicrobial activity. Journal of Saudi Chemical Society, 15(1), 59–65. doi:10.1016/j.jscs.2010.10.006
- Agrawal, M., Kharkar, P., Moghe, S., Mahajan, T., Deka, V., Thakkar, C., … Vishwakarma, R. A. (2013). Discovery of thiazolyl-phthalazinone acetamides as potent glucose uptake activators via high-throughput screening. Bioorganic & Medicinal Chemistry Letters, 23(20), 5740–5743. doi:10.1016/j.bmcl.2013.07.067
- Ahmad, A., Varshney, H., Rauf, A., Husain, F. M., & Ahmad, I. (2014). Synthesis, biological screening of novel long chain derivatives of 1,3-disubstituted-1H-pyrazol-5(4H)-one and 2-substituted-3H-1,4-phthalazin-1,4-dione: Structure-activity relationship studies. Journal of King Saud University – Science, 26(4), 290–299. doi:10.1016/j.jksus.2013.09.003
- Al Mousawi, S. M., EL-Apasery, M. A., & Elnagdi, M. H. (2008). Studies with condensed aminothiophenes: Microwave assisted cycloaddition reactions of thieno[3,4-d]-pyridazinone and thieno[3,4-c]quinolinone. Heterocycles, 75(5), 1151–1161. doi:10.3987/COM-07-11301
- Al Saleh, B., Hilmy, N. M., El-Apasery, M. A., & Elnagdi, M. H. (2006). Microwaves in organic synthesis: Synthesis of pyridazinones, phthalazinones and pyridopyridazinones from 2-oxo-arylhydrazones under microwave irradiation. Journal of Heterocyclic Chemistry, 43(6), 1575–1581. doi:10.1002/jhet.5570430622
- Almahli, H., Hadchity, E., Jaballah, M. Y., Daher, R., Ghabbour, H. A., Kabil, M. M., … El Dehna, W. M. (2018). Development of novel synthesized phthalazinone-based PARP-1 inhibitors with apoptosis inducing mechanism in lung cancer. Bioorganic Chemistry, 77, 443–456. doi:10.1016/j.bioorg.2018.01.034
- Ayyad, R. A., Sakr, H., & El-Gamal, K. (2016). Synthesis, modeling and anticonvulsant activity of some phthalazinone derivatives. American Journal of Organic Chemistry, 6(1), 29–38. doi:10.5923/j.ajoc.20160601.04
- Behalo, M. S., Gad El-Karim, I. A., & Rafaat, R. (2017). Synthesis of novel phthalazine derivatives as potential anticancer and antioxidant agents based on 1-chloro-4-(4-phenoxyphenyl)phthalazine. Journal of Heterocyclic Chemistry, 54(6), 3591–3599. doi:10.1002/jhet.2985
- Boraei, A. T., Ashour, H. K., El-Sayed, H., Abdelmoaty, N., El-Falouji, A. I., & Gomaa, M. S. (2019). Design and synthesis of new phthalazine-based derivatives as potential EGFR inhibitors for the treatment of hepatocellular carcinoma. Bioorganic Chemistry, 85, 293–307. doi:10.1016/j.bioorg.2018.12.039
- Bunce, R. A., Harrison, T., & Nammalwar, B. (2012). Efficient synthesis of selected phthalazine derivatives. Heterocyclic Communications, 18(3), 123–126. doi:10.1515/hc-2012-0079
- Cao, P., Qu, J., Burton, G., & Rivero, R. A. (2008). Facile synthesis of 6-aryl 5-N-substituted pyridazinones: Microwave-assisted Suzuki-Miyaura cross coupling of 6-chloropyridazinones. The Journal of Organic Chemistry, 73(18), 7204–7208. doi:10.1021/jo801097v
- Damera, T., Pagadala, R., Rana, S., & Jonnalagadda, S. B. (2023). A concise review of multicomponent reactions using novel heterogeneous catalysts under microwave irradiation. Catalysts, 13(7), 1034. doi:10.3390/catal13071034
- Daştan, A., Kulkarni, A., & Török, B. (2012). Environmentally benign synthesis of heterocyclic compounds by combined microwave-assisted heterogeneous catalytic approaches. Green Chemistry, 14(1), 17–37. doi:10.1039/C1GC15837F
- Eicher, T., Hauptmann, S., & Speicher, A. (2012). The chemistry of heterocycles: Structures, reactions, synthesis and applications (3rd ed.) Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA (pp. 457–500). https://application.wiley-vch.de/books/sample/3527327479_c01.pdf
- El Adl, K., Ibrahim, M. K., Khedr, F., Abulkhair, H. S., & Eissa, I. H. (2022). Design, synthesis, docking, and anticancer evaluations of phthalazines as VEGFR‐2 inhibitors. Archiv der Pharmazie, 355(1), e2100278. doi:10.1002/ardp.202100278
- El Rayes, S. M., El-Enany, G., Ali, I. A., Ibrahim, W., & Nafie, M. S. (2022). Synthesis of novel phthalazinedione based derivatives with promising cytotoxic, anti-bacterial, and molecular docking studies as VEGFR2 inhibitors. ACS Omega, 7(30), 26800–26811. doi:10.1021/acsomega.2c03182
- El Sayed, D., El Rayes, S. M., Soliman, H. A., AlBalaa, I. E., Alturki, M. S., Al Khzem, A. H., … Nafie, M. S. (2024). Synthesis of novel phthalazine-based derivatives with potent cytotoxicity against HCT-116 cells through apoptosis and VEGFR2 inhibition. RSC Advances, 14(19), 13027–13043. doi:10.1039/d4ra02103g
- El Shamy, I. E., Hleli, E., Alsheikh, A. A., Yawer, M. A., El-Hashash, M. A., Dybal, J., & Abdel-Mohsen, A. M. (2023). Synthesis of some mono- and disaccharide-grafting phthalazine derivatives and some new Se-nucleosideanalogues: Antibacterial properties, quantum chemical calculations, and cytotoxicity. Molecules, 28(1), 317. doi:10.3390/molecules28010317
- Eldehna, W. M., Ibrahim, H. S., Abdel-Aziz, H. A., Farrag, N. N., & Youssef, M. M. (2015). Design, synthesis and in vitro antitumor activity of novel N-substituted-4-phenyl/benzylphthalazin-1-ones. European Journal of Medicinal Chemistry, 89, 549–560. doi:10.1016/j.ejmech.2014.10.064
- Emam, S. M., Rayes, S. M. E., Ali, I. A. I., Soliman, H. A., & Nafie, M. S. (2023). Synthesis of phthalazine‑based derivatives as selective anti‑breast cancer agents through EG FR‑mediated apoptosis: In vitro and in silico studies. BMC Chemistry, 17(1), 90. doi:10.1186/s13065-023-00995-2
- Ezzat, M. A. F., Abdelhamid, S. M., Fouad, M. A., Abdel‐Aziz, H. A., & Allam, H. A. (2023). Design, synthesis, in vitro, and in vivo evaluation of novel phthalazinone‐based derivatives as promising acetylcholinesterase inhibitors for treatment of Alzheimer’s disease. Drug Development Research, 84(6), 1231–1246. doi:10.1002/ddr.22082
- Hameed, A. D., Ovais, S., Yaseen, R., Rathore, P., Samim, M., Singh, S., … Javed, K. (2016). Synthesis and biological evaluation of new phthalazinone derivatives as anti-inflammatory and anti-proliferative agents. Archiv der Pharmazie, 349(2), 150–159. doi:10.1002/ardp.201500336
- Hashash, M. A. E. (2014). Synthesis of novel series of phthalazine derivatives with antibacterial and antifungal evaluation. Journal of Chemical Engineering & Process Technology, 5(4), 4–9. doi:10.4172/2157-7048.1000195
- Henary, M., Kananda, C., Rotolo, L., Savino, B., Owens, E. A., & Cravotto, G. (2020). Benefits and applications of microwave-assisted synthesis of nitrogen containing heterocycles in medicinal chemistry. RSC Advances, 10(24), 14170–14197. doi:10.1039/d0ra01378a
- Ibrahim, H. S., Eldehna, W. M., Abdel-Aziz, H. A., Elaasser, M. M., & Abdel-Aziz, M. M. (2014). Improvement of antibacterial activity of some sulfa drugs through linkage to certain phthalazin-1-(2H)-one scaffolds. European Journal of Medicinal Chemistry, 85, 480–486. doi:10.1016/j.ejmech.2014.08.016
- Inoue, K., Urushibara, K., Kanai, M., Yura, K., Fujii, S., Ishigami-Yuasa, M., … Hirano, T. (2015). Design and synthesis of 4-benzyl-1-(2H)-phthalazinone derivatives as novel androgen receptor antagonists. European Journal of Medicinal Chemistry, 102, 310–319. doi:10.1016/j.ejmech.2015.08.002
- Javahershenas, R., Makarem, A., & Klika, K. D. (2024). Recent advances in microwave-assisted multicomponent synthesis of spiro heterocycles. RSC Advances, 14(8), 5547–5565. doi:10.1039/d4ra00056k
- Joule, J. A., & Mills, K. (2000). Heterocyclic chemistry (5th ed.). New York: John Wiley and Sons (pp. 249–288). https://kgut.ac.ir/useruploads/1615027155168dde.pdf
- Khalifa, M. M., Al-Karmalawy, A. A., Elkaeed, E. B., Nafie, M. S., Tantawy, M. A., Eissa, I. H., & Mahdy, H. A. (2021). Topo II inhibition and DNA intercalation by new phthalazine based derivatives as potent anticancer agents: Design, synthesis, anti-proliferative, docking, and in vivo studies. Journal of Enzyme Inhibition and Medicinal Chemistry, 37(1), 299–314. doi:10.1080/14756366.2021.2007905
- Khidre, R. E., Mostafa, M. S., Mukhrish, Y. E., Salem, M. A., & Behalo, M. S. (2022). Synthetic methods of 1H-pyrazolo[1,2-b]phthalazine derivatives. Current Organic Chemistry, 26(22), 2055–2069. doi:10.2174/1385272827666230124145625
- Kotb, A. R., Bakhotmah, D. A., Abdallah, A. E., Elkady, H., Taghour, M. S., Eissa, I. H., & El-Zahabi, M. A. (2022). Design, synthesis, and biological evaluation of novel bioactive thalidomide analogs as anticancer immunomodulatory agents. RSC Advances, 12(52), 33525–33539. doi:10.1039/d2ra06188k
- Liu, Q., Zhang, B., Wang, Y., Wang, X., & Gou, S. (2022). Discovery of phthalazino[1,2-b]-quinazolinone derivatives as multi-target HDAC inhibitors for the treatment of hepatocellular carcinoma via activating the p53 signal pathway. European Journal of Medicinal Chemistry, 229, 114058. doi:10.1016/j.ejmech.2021.114058
- Lu, D., Liu, J., Zhang, Y., Liu, F., Zeng, L., Peng, R., … Zuo, J. (2018). Discovery and optimization of phthalazinone derivatives as a new class of potent dengue virus inhibitors. European Journal of Medicinal Chemistry, 145, 328–337. doi:10.1016/j.ejmech.2018.01.008
- Majumder, A., Gupta, R., & Jain, A. (2013). Microwave-assisted synthesis of nitrogen containing heterocycles. Green Chemistry Letters and Reviews, 6(2), 151–182. doi:10.1080/17518253.2012.733032
- Marzouk, M. I., Shaker, S. A., Hafiz, A. A., & El-Baghdady, K. Z. (2016). Design and synthesis of new phthalazinone derivatives containing benzyl moiety with anticipated antitumor activity. Biological & Pharmaceutical Bulletin, 39(2), 239–251. doi:10.1248/bpb.b15-00656
- Mourad, A., K., Makhlouf, A. A., Soliman, A. Y., & Mohamed, S. A. (2020). Phthalazines and phthalazine hybrids as antimicrobial agents: Synthesis and biological evaluation. Journal of Chemical Research, 44(1-2), 31–41. doi:10.1177/17475198198838
- Moustafa, A. H., El‐Sayed, H. A., Abd El‐Hady, R. A., Haikal, A. Z., & El‐Hashash, M. (2016). Synthesis, antiviral, and antimicrobial activity of N‐ and S‐alkylated phthalazine derivatives. Journal of Heterocyclic Chemistry, 53(3), 789–799. doi:10.1002/jhet.2316
- Rao, K. P., Basak, A. K., Deb, P. K., Sharma, S., & Reddy, L. K. (2013). Microwave assisted palladium-catalyzed synthesis of phthalazinones and pyridopyridazinones. Tetrahedron Letters, 54(28), 3694–3696. doi:10.1016/j.tetlet.2013.05.006
- Richard, G., Frank, S., Kenneth, W., Humera, A., Lorraine, B., Lena, L., & John, R. (1986). The use of microwave ovens for rapid organic synthesis. Tetrahedron Letters, 27(3), 279–282. doi:10.1016/S0040-4039(00)83996-9
- Romero, A. H., Rodríguez, N., Oviedo, H., & Lopez, S. E. (2019). Antileismanial activity, mechanism of action study and molecular docking of 1,4‐bis (substituted benzalhydrazino) phthalazines. Archiv der Pharmazie, 352(6), e1800299. doi:10.1002/ardp.201800299
- Romero, A. H., Sojo, F., Arvelo, F., Calderón, C., Morales, A., & López, S. E. (2020). Anticancer potential of new 3-nitroaryl-6-(N-methyl)piperazin-1,2,4-triazolo-[3,4-a] phthalazines targeting voltage-gated K+ channel: Copper-catalyzed one-pot synthesis from 4-chloro-1-phthalazinyl-arylhydrazones. Bioorganic Chemistry, 101, 104031. doi:10.1016/j.bioorg.2020.104031
- Rose, M., Burgess, J. T., O'Byrne, K., Richard, D. J., & Bolderson, E. (2020). PARP inhibitors: Clinical relevance, mechanisms of action and tumor resistance. Frontiers in Cell and Developmental Biology, 8, 564601. doi:10.3389/fcell.2020.564601
- Sangshetti, J., Pathan, S. K., Patil, R., Akber Ansari, S., Chhajed, S., Arote, R., & Shinde, D. B. (2019). Synthesis and biological activity of structurally diverse phthalazine derivatives: A systematic review. Bioorganic & Medicinal Chemistry, 27(18), 3979–3997. doi:10.1016/j.bmc.2019.07.050
- Sethy, C., & Kundu, C. N. (2022). PARP inhibitor BMN-673 induced apoptosis by trapping PARP-1 and inhibiting base excision repair via modulation of pol-beta in chromatin of breast cancer cells. Toxicology and Applied Pharmacology, 436, 115860. doi:10.1016/j.taap.2021.115860
- Siraj, A. K., Pratheeshkumar, P., Parvathareddy, S. K., Divya, S. P., Al-Dayel, F., Tulbah, A., … Al-Kuraya, K. S. (2018). Overexpression of PARP is an independent prognostic marker for poor survival in Middle Eastern breast cancer and its inhibition can be enhanced with embelin co-treatment. Oncotarget, 9(99), 37319–37332. doi:10.18632/oncotarget.26470
- Sroczyński, D., Malinowski, Z., Szcześniak, A. K., & Pakulska, W. (2016). New 1(2H)-phthalazinone derivatives as potent nonpeptidic HIV-1 protease inhibitors: Molecular docking studies, molecular dynamics simulation, oral bioavailability and ADME prediction. Molecular Simulation, 42(8), 628–641. doi:10.1080/08927022.2015.1067808
- Tantawy, M. A., Shalby, A. B., Barnawi, I. O., Kattan, S. W., Abd-Rabou, A. A., & Elmegeed, G. A. (2023). Anti-cancer activity, and molecular docking of novel hybrid heterocyclic steroids revealed promising anti-hepatocellular carcinoma agent: Implication of cyclin dependent kinase-2 pathway. Steroids, 193, 109187. doi:10.1016/j.steroids.2023.109187
- Teran, C., Besada, P., Vila, N., Costas-Lago, M. C., & Ter, C. (2019). Recent advances in the synthesis of phthalazin-1-(2H)-one core as a relevant pharmacophore in medicinal chemistry. European Journal of Medicinal Chemistry, 161, 468–478. doi:10.1016/j.ejmech.2018.10.047
- Vila, N., Besada, P., Brea, J., Loza, M. I., & Terán, C. (2022). Novel phthalazin-1(2H)-one derivatives displaying a dithiocarbamate moiety as potential anticancer agents. Molecules, 27(23), 8115. doi:10.3390/molecules27238115
- Vila, N., Besada, P., Costas, T., Costas-Lago, M. C., & Terán, C. (2015). Phthalazin-1(2H)-one as a remarkable scaffold in drug discovery. European Journal of Medicinal Chemistry, 97(1), 462–482. doi:10.1016/j.ejmech.2014.11.043
- Wasfy, A. F., Aly, A. A., Behalo, M. S., & Mohamed, N. S. (2020). Synthesis of novel phthalazine derivatives as pharmacological activities. Journal of Heterocyclic Chemistry, 57(1), 12–25. doi:10.1002/jhet.3735
- Zhang, S., Zhao, Y., Liu, Y., Chen, D., Lan, W., Zhao, Q., … Gong, P. (2010). Synthesis and antitumor activities of novel 1,4-disubstituted phthalazine derivatives. European Journal of Medicinal Chemistry, 45(8), 3504–3510. doi:10.1016/j.ejmech.2010.05.016