
Abstract
Autoinflammatory diseases are characterized by recurrent systemic inflammation due to dysfunction of the innate immune system, and they are originally the hereditary recurrent fever syndromes that develop in early childhood. Many cases are thus diagnosed in the pediatric field, but there are many cases that are not diagnosed until adulthood, including adult-onset cases. Accordingly, not only pediatricians but also rheumatologists and primary care physicians play a major role in the diagnosis and treatment of autoinflammatory diseases. Broad non-hereditary autoinflammatory diseases such as adult-onset Still disease and Behcet's disease are also often encountered in routine internal medicine practice. With recent developments in genetic testing, the conditions of patients who have been diagnosed and treated as having fevers of unknown etiology have been increasingly classified as autoinflammatory diseases. Regarding treatment in Japan, biological agents (such as canakinumab) that inhibit IL-1β are now administered to patients with autoinflammatory diseases. In this review, we introduce an approach from rheumatologists based on diagnoses and treatment of ‘classical” autoinflammatory diseases in adults, focusing on familial Mediterranean fever, which has a high frequency in adults.
1. Introduction
Autoinflammatory diseases are characterized by a recurrent fever of unknown cause and systemic inflammation [Citation1]. The pathological condition of autoinflammatory disease is mainly abnormal function in cells of the innate immune system such as monocytes, macrophages, neutrophils and natural killer (NK) cells [Citation2]. Unlike autoimmune diseases underlying the dysfunction of adaptive immunity, autoinflammatory diseases are less closely related to HLA class II, and autoantibodies are usually not detected in patients with autoinflammatory diseases. Kastner, therefore, defines autoinflammation as cases in which: (1) an inflammatory finding for which the trigger is not clear is present, (2) there is no association with a high titer of autoantibodies or self-responsive T cells and (3) an abnormality of innate immunity is observed [Citation1].
The spectrum of autoinflammatory diseases has been extended to not only the hereditary recurrent fever syndromes, including familial Mediterranean fever (FMF), tumor necrosis factor (TNF)-related periodic fever syndrome (TRAPS) and cryopyrin-associated periodic syndrome (CAPS), but also uncertain genetic etiologies including adult-onset Still disease, Behcet's disease and gout [Citation3]. Since hereditary autoinflammatory diseases usually develop in early childhood, many cases are diagnosed in the pediatric field. However, because of recent advances in genetic diagnoses as well as cases diagnosed for the first time in adulthood, it is apparent that rheumatologists and physicians can play a major role in the diagnosis and treatment of autoinflammatory diseases.
In this review, we highlight the pathology, diagnosis and treatment of autoinflammatory diseases (focusing on FMF), and we discuss the role of rheumatologists in the diagnosis and treatment of autoinflammatory diseases in adults.
1.1. Pathology of familial Mediterranean fever
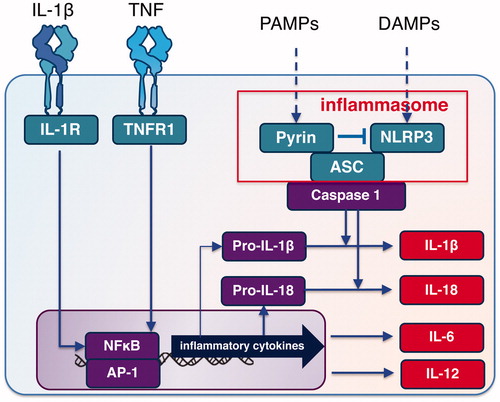
Inflammation in autoinflammatory diseases is caused by an activation of antigen-independent immune cells (monocytes, macrophages, neutrophils and NK cells). Many of the narrowly defined hereditary autoinflammatory diseases exhibit recurrent fever and are called periodic fever syndromes [Citation4]. The inflammasome is a multiprotein oligomer responsible for the activation of inflammatory responses activated by various stimuli including pathologies, bacteria, microbial toxins, xenobiotics, PAMPs (pathogen-associated molecular patterns) and DAMPS (damage-associated molecular patterns).
FMF is characterized by recurrent fever by arthritis and serositis, and polymorphisms or mutations of Mediterranean fever (MEFV) gene encoding pyrin are closely related to the pathogenesis as a disease susceptibility gene for FMF [Citation5]. shows the clinical manifestations of FMF.
Table 1. Clinical manifestations of FMF.
Since pyrin is involved in the negative regulation of NLRP3 inflammasome, dysfunction of pyrin results in an increased production of inflammatory cytokines, including interleukin (IL)-lβ and IL-18 by the activation of NLRP3 inflammasome [Citation6]. It has also been suggested that the activation of pyrin inflammasome by a function-acquired pyrin mutation also contributes to the pathology of FMF [Citation7].
On the other hand, activation of the NFκB signaling pathway (which increases the amounts of TNF-α and IL-6) is important for the production of IL-1β precursor and IL-18 precursor as the first signal for inflammasome activation [Citation8] (). Accordingly, the blockage of these inflammatory cytokines with biologic agents is considered to be a reasonable approach for the treatment of FMF [Citation9].
Figure 1. Inflammasome and cytokines in FMF. ASC: apoptosis-associated speck-like protein containing a caspase-recruitment domain; DAMPs: damage-associated molecular patterns; IL-1R: interleukin-1beta receptor; NLRP3: Nod-like receptor protein 3; PAMPs: pathogen-associated molecular patterns; TNFR: tumor necrosis factor receptor.
1.2. Diagnosis of FMF in adults
Among the hereditary periodic fever syndromes, FMF is the most common disease diagnosed in adults, whereas TRAPS and CAPS are extremely rare [Citation10]. FMF is estimated to affect approx. 1000–2000 patients in Japan, but both the clinical manifestations and the pattern of gene mutation/polymorphism in Japanese are different from those observed in the Mediterranean coastal region and Middle East where the incidence of FMF is high among Jewish, Turkish, Armenian populations. Japanese FMF patients had a lower percentage of MEFV mutations in exon 10, and FMF cases in Japan have been reported to be more frequently adult-onset [Citation11].
Although there are a number of mutations/variants in MEFV gene among FMF patients, a clinical diagnosis is the gold standard for FMF [Citation12]. In Japan, we usually use the FMF diagnosis flowchart provided by the Ministry of Health, Labor and Welfare research project ‘Diagnostic criteria for auto-inflammatory diseases and related diseases, severity classification, and study on the establishment of clinical practice guidelines’ compiled by Professor Heike's group [Citation13] (). When a rheumatologist or physician diagnoses adult-onset FMF, the following items should be considered: (1) the patient's medical history, (2) the interpretation of MEFV genetic diagnosis and (3) differentiation from other collagen diseases.
Figure 2. Flowchart for diagnostic evaluation of FMF.
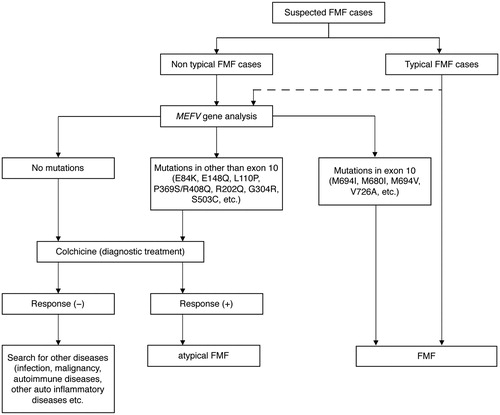
Regarding the patient's medical history, it is important to check not only present and past symptoms (including fever of periodicity and abdominal pain) in the patient's childhood but also the family history including the patient's children. For the interpretation of the MEFV gene findings, special attention is required for an E148Q heterozygous mutation in exon 2, which is a common polymorphism in Japanese. According to the Tohoku genome database (http://ijgvd.megabank.tohoku.ac.jp/), the Japanese allele frequency of E148Q (rs3743930) is 21.57%. In addition, since 10%–20% of patients who meet the clinical diagnostic criteria of FMF do not carry any known mutation for FMF, it is not possible to use only genetic testing for the exclusion of diagnoses.
Although the differential diagnosis of FMF in adults varies with the patient's predominant clinical features, it is sometimes difficult to distinguish FMF from other rheumatic disorders, especially in elderly-onset FMF. In such cases, it might be useful to check the results of genetic testing, the duration of fevers, the distribution of arthritis, the presence or absence of hyperferritinemia in the serum and the serum level of IgD.
1.3. Treatment of FMF in adults
The therapeutic goal for FMF patients is to suppress chronic inflammation due to periodic fever attacks and to prevent secondary amyloidosis, which can cause a significant decrease in the patient's quality of life and irreversible organ damage. For physicians, the prevention, early diagnosis and appropriate treatment of amyloidosis in adult FMF patients are important factors affecting the long-term prognosis.
Colchicine is used as a prophylactic treatment for FMF attacks, and it is recommended in all FMF patients regardless of the frequency and intensity of attacks [Citation14]. The use of intermittent high-dose colchicine only during attacks cannot prevent the onset of amyloidosis caused by chronic inflammation [Citation15,Citation16]. In addition, some effects of an IL-1β inhibitor on amyloidosis have been shown [Citation17], but the evidence for this treatment is not sufficient at present. Because IL-6 inhibitors can dramatically inhibit the production of serum amyloid A protein, they may have greater efficacy in the treatment of amyloidosis. Indeed, the effectiveness of IL-6 inhibitors against amyloidosis for severe FMF has been reported [Citation18] and has attracted attention as a new therapeutic target.
2. Conclusions
We have briefly outlined the diagnosis and treatment of FMF from a rheumatologist's viewpoint, which is that FMF is an autoinflammatory disease relatively rarely encountered in adults. Autoinflammatory diseases are a new disease concept, and elucidation of the disease state is under development. With the progress of genetic diagnostic techniques in recent years and the use of biological agents, the diagnostic ability and the prognosis of autoinflammatory diseases have improved. In the future, it is necessary to determine the relationship between the genetic features of autoinflammatory diseases including FMF and each disease type and clinical symptom, and to elucidate biomarkers for predicting the therapeutic response.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- McDermott MF, Aksentijevich I, Galon J, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 1999;97:133–144.
- de Jesus AA, Canna SW, Liu Y, et al. Molecular mechanisms in genetically defined autoinflammatory diseases: disorders of amplified danger signaling. Annu Rev Immunol. 2015;33:823–874.
- Federici S, Sormani MP, Ozen S, et al. Evidence-based provisional clinical classification criteria for autoinflammatory periodic fevers. Ann Rheum Dis. 2015;74:799–805.
- Kastner DL. Hereditary periodic fever syndromes. Hematology Am Soc Hematol Educ Program. 2005;2005:74–81.
- Stehlik C, Reed JC. The PYRIN connection: novel players in innate immunity and inflammation. J Exp Med. 2004;200:551–558.
- Kim ML, Chae JJ, Park YH, et al. Aberrant actin depolymerization triggers the pyrin inflammasome and autoinflammatory disease that is dependent on IL-18, not IL-1β. J Exp Med. 2015;212:927–938.
- Chae JJ, Cho YH, Lee GS, et al. Gain-of-function Pyrin mutations induce NLRP3 protein-independent interleukin-1β activation and severe autoinflammation in mice. Immunity. 2011;34:755–768.
- Bagci S, Toy B, Tuzun A, et al. Continuity of cytokine activation in patients with familial Mediterranean fever. Clin Rheumatol. 2004;23:333–337.
- Koga T, Migita K, Kawakami A. Biologic therapy in familial Mediterranean fever. Mod Rheumatol. 2016;26:637–641.
- Yao Q, Lacbawan F, Li J. Adult autoinflammatory disease frequency and our diagnostic experience in an adult autoinflammatory clinic. Semin Arthritis Rheum. 2016;45:633–637.
- Migita K, Agematsu K, Yazaki M, et al. Familial mediterranean fever: genotype-phenotype correlations in Japanese patients. Medicine (Baltimore). 2014;93:158–164.
- Giancane G, Ter Haar NM, Wulffraat N, et al. Evidence-based recommendations for genetic diagnosis of familial Mediterranean fever. Ann Rheum Dis. 2015;74:635–641.
- Pediatric Rheumatology association of Japan. Autoinflammatory disease clinical practice guideline 2017. Tokyo: Shindan To Chiryo Sha; 2017.
- Goldfinger SE. Colchicine for familial Mediterranean fever. N Engl J Med. 1972;287:1302.
- Ozen S, Demirkaya E, Erer B, et al. EULAR recommendations for the management of familial Mediterranean fever. Ann Rheum Dis. 2016;75:644–651.
- Wright DG, Wolff SM, Fauci AS, et al. Efficacy of intermittent colchicine therapy in familial Mediterranean fever. Ann Intern Med. 1977;86:162–165.
- Sevillano AM, Hernandez E, Gonzalez E, et al. Anakinra induces complete remission of nephrotic syndrome in a patient with familial mediterranean fever and amyloidosis. Nefrologia. 2016;36:63–66.
- Yilmaz S, Cinar M, Simsek I, et al. Tocilizumab in the treatment of patients with AA amyloidosis secondary to familial Mediterranean fever. Rheumatology (Oxford). 2015;54:564–565.