
Abstract
Recent studies have revealed a relationship between cellular metabolism and cell function in immune cells. Cellular metabolism not only provides supplemental ATP, but also supports dynamic changes in cell proliferation and differentiation. For example, T cells exhibit subset-specific metabolic profiles, and require certain types of metabolism for their functions. Determining the metabolic profiles that support inflammatory immune responses may lead to novel treatment strategies for chronic inflammatory diseases such as rheumatoid arthritis (RA). However, the mechanisms by which metabolism modulates cell function have been unclear. Recent studies have begun to unveil unexpected non-metabolic functions for metabolic enzymes in the context of inflammation, including roles in signaling and gene regulation. Here we describe recent findings related to immunometabolism, the metabolome of RA patients, and the metabolically independent functions of glycolytic enzymes. We discuss how metabolic processes impact immune cells, especially T cells and fibroblast like synoviocytes, which are considered the orchestrators of autoimmune arthritis.
1. Introduction
Warburg et al. performed pioneering research on respiration and biosynthesis in the early part of the 20th century. In the 1920s, he presented the first evidence that cancer cells produce lactate from glucose under normoxic conditions [Citation1]. This phenomenon is referred to as the ‘Warburg effect.’ Recently, many studies have examined the modulation of cancer cell metabolism as a therapeutic strategy in animal models [Citation2].
Like cancer cells, immune cells use different metabolic pathways to support their differentiation and phenotype maintenance [Citation3]. Naïve T cells in quiescence are fueled by fatty acid oxidation (FAO). Interferon-γ (IFN-γ)-producing helper T lymphocytes (Th1 cells) require an increase in glycolysis for their activation and IFN-γ production [Citation4]. For their differentiation, interleukin-17 (IL-17)-producing helper T lymphocytes (Th17 cells) depend mainly on glycolysis, while regulatory T lymphocytes (Treg cells) rely on FAO and oxidative phosphorylation (OXPHOS). This metabolic repertoire associated with immune cell differentiation and function is now called immunometabolism. In addition, fibroblasts are reported to have an altered metabolism in patients with rheumatoid arthritis (RA) [Citation5].
In recent years, substantial findings have been revealed in the area of metabolic reprogramming. Here we provide a brief review of the major metabolic pathways involved in RA, particularly glycolysis, FAO, and OXPHOS.
2. Metabolomics in rheumatoid arthritis
Young and colleagues first reported in 2013 that the metabolism was altered in the serum of disease-modifying antirheumatic drug (DMARD)-naïve RA patients [Citation6]. They used nuclear magnetic resonance spectroscopy-based metabolomics to evaluate the serum from patients with established RA, those with early arthritis, and healthy controls. Many metabolites were correlated with the serum C-reactive protein (CRP) level. The metabolites of glycolytic pathways (glucose, lactate) were increased in the active RA patients. Lipid metabolites were also associated with serum CRP levels, and might contribute to the increased atherosclerosis associated with inflammatory disease. In addition, Lauridsen and co-workers [Citation7] determined the metabolic profiles in the plasma of RA patients with active synovitis versus those in remission. They analyzed two patient groups: (i) 27 patients in remission treated with anti-tumor necrosis factor α inhibitors (TNFi) and conventional synthetic DMARDs, and (ii) 21 symptomatic patients treated with conventional synthetic DMARDs only (11 patients received methotrexate (MTX), 5 received salazosulfapyridine (SSZ), and 6 received glucocorticoid (GC); none of these patients received TNFi). These authors showed that the metabolomes from RA patients with active synovitis were different from those of patients in remission. However, notably, the metabolic profiles of symptomatic patients were not significantly different from those of the patients in remission after the former received an optimized therapy (including TNFi). Furthermore, the metabolic fingerprints of both groups of RA patients were different from those of healthy subjects. These findings suggested that none of the treatments truly cures RA, as the underlying pathology of the RA was not fully reversed. Several other reports also demonstrated metabolic alterations in RA patients [Citation8–12] (). These data suggested that metabolic changes are closely associated with inflammation in RA patients and may represent a potential target for disease monitoring and personalized medication for RA. However, further evaluation with larger numbers of patients in a multicenter setting is still required before these approaches can be realized.
Table 1. Metabolomic differences between RA patients and healthy subjects.
3. Limiting enzymes involved in the Warburg effect
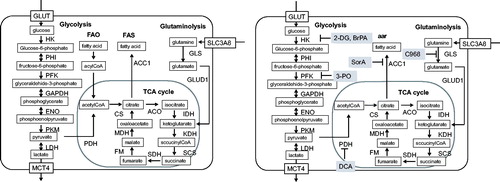
There are a number of rate-limiting enzymes in cellular metabolism (). Glycolysis refers to a sequence of cytosolic enzymatic reactions that convert glucose into pyruvate, generating energy. In general, glucose is imported into the cytosol by glucose transporter-1 (GLUT1). The glucose is then initially converted to glucose-6-phosphate by the rate-limiting enzyme hexokinase (HK), and subsequently to fructose-6-phosphate spontaneously. A second rate-limiting enzyme, phosphofructokinase-1 (PFK1), enables the formation of fructose-1, 6-bisphosphate. Fructose-1, 6-bisphosphate is then converted to phosphoenolpyruvate by several steps. Pyruvate is then formed from phosphoenolpyruvate by another rate-limiting enzyme, pyruvate kinase muscle isozyme (PKM). The cytosolic pyruvate has two alternative routes for catabolism. When oxygen is abundant, pyruvate dehydrogenase (PDH) transforms pyruvate to Acetyl-CoA. Acetyl-CoA can enter the tricarboxylic acid (TCA) cycle to produce NADH and FADH, which can generate ATP by OXPHOS. One glucose molecule metabolized by aerobic glycolysis can generate two ATP molecules. In contrast, if the products generated by glycolysis can be further metabolized by the TCA cycle and OXPHOS, the theoretical maximum number of ATP molecules from one molecule of glucose is 32. The second fate of pyruvate is its conversion to lactate in the cytosol by lactate dehydrogenase (LDH); lactate is then transported to the extracellular space by monocarboxylate transporter (MCT). Lactate generation occurs under hypoxic conditions. This process generates NAD+, which is consumed during glycolysis. When cells cannot regenerate NAD + by OXPHOS, the strategy of generating lactate can provide NAD + and remove the cells’ limitation to glycolysis. Cancer cells and some immune cells still use glucose to generate lactate despite the presence of abundant oxygen, a phenomenon known as the ‘Warburg effect’ [Citation1,Citation3]. Although the requirement for mitochondrial ATP production is decreased in tumor cells, the demand for biosynthetic precursors and NADPH is increased. To compensate for these changes and to maintain a functional TCA cycle, cancer cells often rely on elevated glutaminolysis [Citation13]. This use of glutaminolysis to compensate for glycolysis is not only tightly linked to different biosynthetic pathways but also generates the anaplerotic substrate α-ketoglutarate (α-KG), which can be metabolized through the TCA cycle to generate either citrate or pyruvate. Notably, glycolysis, the TCA cycle, and glutaminolysis not only provide energy but also generate many intermediates for other metabolic pathways, such as the pentose phosphate pathway (PPP), hexosamine biosynthesis, amino acid biosynthesis, and de novo fatty acid synthesis (FAS).
Figure 1. Metabolome maps showing the key enzymes for each pathway and specific inhibitors.
4. Cellular metabolism in immune cells
The altered metabolism in RA mentioned above is important for understanding RA’s pathology. Although metabolomics is the systematic study of chemical fingerprints, immune-cell metabolism was first studied independently of metabolomics. Cooper et al. [Citation14] first reported in the 1960s that activated human lymphocytes have an increased demand for ATP. Since then, many studies have analyzed the detailed mechanism of T cell activation with respect to cellular metabolism [Citation3] (). Naïve T cells in quiescence require only housekeeping ATP production, and resting cells generate 96% of their ATP by OXPHOS, and only 4% by glycolysis [Citation15]. When T cells receive CD3/CD28 stimulation, they initiate protein synthesis and oxygen use for maximal energy extraction, because they grow to approximately double their resting size, enter a rapid-proliferation program, and differentiate from quiescent cells to effector T cells that secrete high levels of cytokines [Citation16]. For these increased energy demands, T cells in activation increase their glycolysis by expressing increased GLUT1 levels and consuming more oxygen [Citation17–19]. OXPHOS produces ATP more efficiently than does glycolysis. Thus, Th1 cells mainly use OXPHOS rather than glycolysis for their proliferation and survival [Citation4,Citation20,Citation21]. However, in the maturation of Th1 cells, glycolysis is important, because it is needed for IFN-γ translation. GAPDH can bind to the IFN-γ mRNA and inhibits its translation, and GAPDH’s binding to glyceraldehyde-3-phosphate enables the translation of IFN-γ [Citation4,Citation20]. Th17 and Treg cells also have unique cellular metabolisms. Th17 cells express high levels of the glycolytic pathway components, including the transporters GLUT1 and MCT4, and the enzymes hexokinase-2 (HK2), PKM, and LDH [Citation22]. In particular, HK2 is an inducible isoform of hexokinase, and is mainly used when increased glycolysis is needed for ATP production. HK2 can be up-regulated approximately 100-fold only in Th17 cells, and thus is considered a key regulator of the glycolysis in these cells. In addition to glycolysis, Th17 cells exhibit increased glutaminolysis and FAS [Citation23,Citation24]. On the other hand, unlike quiescent naïve T cells, Treg cells proliferate continuously at moderate levels, and unlike activated naïve T cells, Th1, and Th17 cells, they do not exhibit a sudden proliferation burst. Treg cells depend only on FAO, and not on OXPHOS or glycolysis [Citation25], although the reason for this dependence is unknown. In addition to ATP production, Treg cells also differ from Th17 cells in their minimal need for amino acid metabolism. Indeed, a depletion of extracellular amino acids suppresses Th17 cell differentiation, while favoring Treg cell development and T cell anergy [Citation26].
Table 2. Metabolic profiles of T cells and dendritic cells.
A recent report suggested that DC activation also requires an increase in glycolysis [Citation27], while FAO is important in tolerogenic DCs (tol-DCs) [Citation28,Citation29]. It is notable that both Th17 cells and activated DCs, which induce immune responses, are dependent on glycolysis. These various metabolic profiles in effector cells are referred to as ‘immunometabolism,’ and are currently of interest as potential therapeutic targets in rheumatic diseases [Citation30,Citation31].
5. T cell metabolism in rheumatoid arthritis
Chronic stimulation and the synovial microenvironment alter the T cell metabolism in RA. Pro-inflammatory cytokines are involved in the pathogenesis of RA. In particular, IL-17 induces stromal cells to produce IL-6 and TNF–α, macrophage activation, and osteoclast differentiation. Thus, IL-17 aggravates synovial inflammation and promotes cartilage and bone destruction [Citation32,Citation33]. In contrast, Foxp3 + regulatory T (Treg) cells promote immune tolerance and inhibit autoimmunity [Citation34]. Thus, the IL-17-producing helper T (Th17)/Treg balance plays an important part in RA development [Citation35].
The cellular metabolism of the T cells in RA patients is not well known. Only a few studies have analyzed the cellular metabolism in peripheral T cells. The peripheral naïve CD4+ T cells of RA patients have a reduced expression of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) [Citation36]. This enzyme is a key regulator of fructose-2, 6-bisphospate, the allosteric activator of PFK, and decreased PFKFB3 levels cause lower glycolysis while increasing the flux to the PPP and the generation of NADPH. Elevated NADPH can neutralize ROS, which, although damaging at high concentrations, are otherwise essential for promoting T cell activation [Citation37]. Indeed, the restoration of T cell ROS can suppress synovial inflammation [Citation36]. In addition to the direct changes in T cells, the hypoxic environment in the RA synovium [Citation38] creates a situation similar to chronic mitochondrial hyperpolarization. That is, the formation of the synovial pannus restricts the availability of oxygen to infiltrating immune cells, which can help alter the glucose and mitochondrial metabolism [Citation39]. This situation is the opposite of that described above, in which naïve T cells mainly rely on OXPHOS for their proliferation and survival, while Th17 cells and Th1 cells require glycolysis for their immune functions. However, there are no reports on the cellular metabolism of Th17 cells and Treg cells from RA patients. While T cell subsets are known to have specific cellular metabolic profiles, which are closely associated with their development and effector functions [Citation4,Citation39], further investigation is required to understand the immunometabolism in RA pathogenesis.
6. Fibroblast like synovial cell metabolism in rheumatoid arthritis
Fibroblast-like synoviocytes (FLSs) are a key component of the invasive synovium and have a major role in the initiation and perpetuation of destructive joint inflammation [Citation40]. FLSs normally assure the structural and dynamic integrity of joints by controlling the composition of the synovial fluid and the extracellular matrix of the joint lining. In RA, however, FLSs display pathogenic properties. RA-FLSs proliferate and become a prominent component of the destructive pannus that characterizes the synovitis in RA patients. Furthermore, RA-FLSs acquire an aggressive phenotype and mediate inflammation and the destruction of the joint. RA-FLSs have a high demand for ATP for their aggressive proliferation during the disease progression (). Like Th17 cells, RA-FLSs have increased expressions of GLUT-1, HK2, pyruvate kinase M2 (PKM2), PFKFB3, and MCT4 [Citation41]. Metabolomics studies also revealed that RA-FLSs have greatly increased glycolysis for ATP production [Citation9,Citation42,Citation43]. While effector T cells require increased glycolysis for their development and effector functions, RA-FLSs require glycolysis and glutaminolysis for their proliferation, survival, activation, and secretion of inflammatory cytokines including chemokines (such as IL6 and MMP-3) [Citation6,Citation41,Citation44].
7. Metabolic alteration as a therapeutic target in experimental models
Many metabolic inhibitors have been shown to have therapeutic effects in animal arthritis models (). A number of glycolysis rate-limiting enzymes are reported to serve as therapeutic targets. We previously reported that inhibiting HK2 with 3-bromopyruvate (BrPA) can transform Th17 cells to Treg cells after their stimulation in vitro (Th17 conditions: IL6, TGF-β, anti-IL4 Ab, anti-IFN-γ Ab, IL-2), and ameliorated the experimental arthritis in SKG mice [Citation45] (). In addition, Panneton et al. [Citation46] showed that BrPA decreased the disease activity in a collagen-induced arthritis (CIA) model. Bian et al. [Citation47] reported that inhibiting PDK with dichloroacetate (DCA) also ameliorated the arthritis in CIA mice.
Figure 2. Metabolic inhibitors affect cell differentiation and activation in the SKG mouse arthritis model.
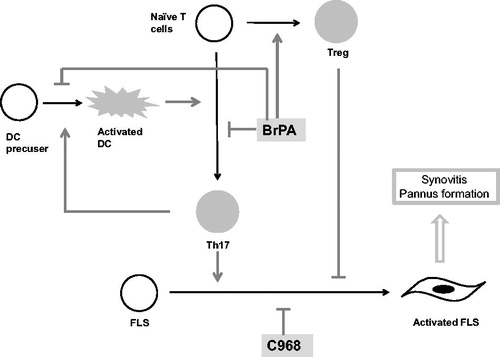
Table 3. Molecules with therapeutic effects in arthritis models
Like T cells, FLSs exhibit increased glycolysis and are a prominent target in arthritis treatment strategies. Blocking HK with 2-DG and BrPA ameliorated the arthritis in CIA mice by blocking FLS proliferation [Citation48].
In the Warburg effect, glycolysis increases and OXPHOS decreases, leading to NADH deficiency, and cells compensate for the lack of NADH by glutaminolysis. However, glutaminolysis is little investigated as a therapeutic target in arthritis. Only Takahashi, our colleague, reported that inhibiting glutainase-1 suppresses the RA-FLS proliferation in vitro and ameliorates the arthritis in SKG mice [Citation41].
Interestingly, Th17 cells have increased FAS, and inhibiting the FAS with soraphen A (SorA) suppresses Th17 cell differentiation and promotes Treg cell development, thus relieving EAE. FAS does not produce ATP or NADH, so it is unclear why SorA suppresses Th17 cells [Citation25].
Our results in SKG mice clearly indicated that metabolic pathways can serve as a target for treating chronic arthritis (). In addition, metabolism-altering nutrition-based treatments are currently being tested in clinical trials (ClinicalTrials.gov Registration Number: NCT02941055.).
7.1. Non-metabolic roles of metabolic enzymes
The mechanism by which glycolysis inhibition suppresses Th17 cells and promotes Treg cells has been extensively investigated. Shi et al. demonstrated that Th17 cell differentiation requires IL-6 stimulation, which activates mTOR, leading to the overexpression of HIF-1α and glycolytic enzymes, and to an increase in glycolytic activity [Citation23]. These findings supported the notion that the up-regulation of T-cell glycolysis is not just a consequence of differentiation, but rather is a necessary step to facilitate differentiation [Citation49]. In addition, several signals, such as ROS, mTOR, PIK3, akt, and HIF-1α, have been found to induce an increase in glycolysis in T cells, but these upstream signals cannot explain how inhibiting glycolysis facilitates the differentiation of Treg cells [Citation23,Citation24,Citation50–52].
Recent studies have started to reveal unexpected non-metabolic functions for metabolic enzymes in the context of inflammation, including roles in signaling and gene regulation (). It is possible that information gained by studying immunometabolism can be leveraged for therapeutic benefit by exploiting these non-canonical features of the metabolic machinery, thus modulating their contribution to the immune response without impacting their basal metabolic functions. In yeast, glucose triggers a broad transcriptional program that suppresses the expression of genes not required for growth in glucose, commonly referred to as ‘glucose repression.’ Deleting HK2 relieves glucose repression [Citation53], while over-expressing GLUT1 cannot reverse glucose repression. However, glucose repression does not require the metabolic function of HK2 [Citation54]. Instead, HK2 binds directly to the transcriptional coactivator Mig1, and in this complex, binds to the promoters of target genes [Citation55,Citation56]. Everts et al. reported that DC activation requires an increase in glycolysis, but not in the ATP from glycolysis or in FAO. Moreover, only inhibiting HK2, and not PFKFB3 or LDH-A, suppressed DCs [Citation28]. These results suggested that HK2 is essential for cell activation through its non-glycolytic function.
Figure 3. Non-enzymatic functions of glycolytic enzymes.
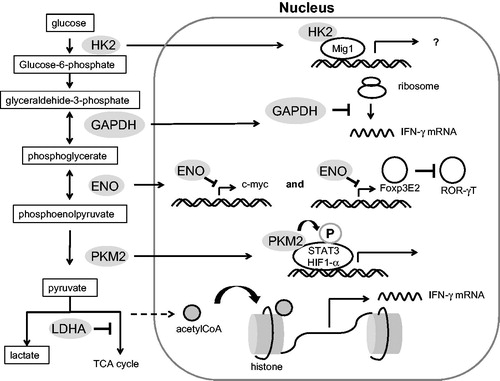
PKM2 can exist as a tetramer or as a dimer that lacks activity as a glycolytic enzyme, and the PKM2 dimer can localize to the nucleus [Citation57]. PKM2 binds to HIF1-α as a coactivator [Citation58]. This activation of HIF1-α does not require the catalytic activity of PKM2, but requires PKM2’s hydroxylation at Pro403 and Pro408 by proline hydroxylase-3 [Citation59]. In addition, PKM2 can directly phosphorylate STAT3. Interestingly, phosphoenol pyruvate is not just a substrate for PKM2, but is also required for STAT3’s phosphorylation by PKM2. STAT3’s phosphorylation requires phosphoenol pyruvate, but not ATP as a phosphate source [Citation60,Citation61].
When the glucose concentration is low, GAPDH binds to AU-rich elements in the 3′-untranslated region (UTR) of mRNAs, including those encoding interferon gamma (IFN-γ) and IL-2 [Citation5,Citation21].
In glycolysis, α-enolase catalyzes the conversion of 2-phosphoglycerate to phosphoenolpyruvate (PEP). In addition, α-enolase is known as Myc promoter binding protein 1 (MBP-1), and it suppresses the pro-proliferative transcription factor c-myc [Citation62,Citation63]. A recent study revealed that Treg cells express high levels of MBP-1. Moreover, enolase-1 directly hampered the transcription of the mRNAs of Foxp3 splicing variants containing exon 2 (FoxP3E2) in induced Treg cells [Citation64]. Foxp3E2 directly inhibited RAR-related orphan receptor gamma T (RORγT), which is an essential transcription factor in Th17 cells. Foxp3E2 directly binds to RORγT via its E2 region [Citation65].
LDH is a tetrameric enzyme variably composed of A and B subunits that, when combined, form a complex with the capability of converting pyruvate to lactate. Peng and colleagues recently showed that T cells mainly express the A subunits of LDH, and that LDH-A is critical for the production of IFN-γ [Citation66]. Depleting LDH-A resulted in the consumption of glycolysis-derived acetyl-CoA, and thus decreased the intracellular acetyl-CoA. Intracellular acetyl-CoA is required for the acetylation that opens the IFN-γ locus during T cell activation [Citation66]. Moreover, LDH-A is reported to bind to 3’AU-rich elements in the GM-CSF mRNA [Citation67].
In this review, we have summarized the recent progress in our understanding of the metabolic changes in immune cells and RA-FLSs in RA, and have also described signaling functions of metabolic enzymes that are distinct from their enzymatic activity. Surprisingly, these transcriptional activities of metabolic enzymes are highly conserved from yeast to humans, and much progress has been made in this field by studying simple model systems. Further exploration of the unique ways in which metabolic processes contribute to immune responses may clarify the relationship between metabolism and inflammation, and lead to new therapeutic strategies for RA.
A. Metabolome map including glycolysis, fatty acid oxidation, fatty acid synthesis, glutaminolysis, and the TCA cycle. B. Molecules with inhibitory effects on metabolic enzymes are indicated by gray shading. FAO: fatty acid oxidation, FAS: fatty acid synthesis, GLUT: glucose transporter, MCT4: monocarboxylate transporter 4, HK: hexokinase, PHI: phosphohexose isomerase, PFK: phosphofructokinase, GAPDH: glyceraldehide-3-phosphate dehydrogenase, ENO: enolase. PKM: pyruvate kinase muscle isozyme, LDH: lactate dehydrogenase, PDH: pyruvate dehydrogenase, CS: citrate synthase, ACO: aconitase, IDH: isocitrate dehydrogenase, KDH: ketoglutarate dehydrogenase, SCS: succinyl CoA synthetase, SDH: succinate dehydrogenase, FM: fumarase, MDH: malate dehydrogenase, SCL3A8: sodium-coupled neutral amino acid transporter 1, GLUD1: glutamate dehydrogenase 1, GLS: glutaminase, ACC1: acetyl-CoA carboxylase-1, 2-DG: 2-Deoxy-D-glucose, BrPA: 3-bromopyruvate, 3-PO: 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one, DCA: diachloroacetate, SorA: soraphen A, DON: 6-diazo-5-oxo-L-norleucine.
Naïve T cells in quiescence rely mainly on FAO for ATP production. In activated naïve T cells, glycolysis and oxidative phosphorylation (OXPHOS) are increased. Effector T cells have various metabolic profiles. Th17 cells rely on glycolysis and glutaminolysis, and have decreased OXPHOS and FAO. FAS is also increased in Th17 cells. Treg cells depend on FAO. Glycolysis is essential for DC activation, while tolerogenic DCs rely on OXPHOS and FAO.
BrPA suppressed Th17 cell differentiation, induced Treg development, and suppressed DC activation, leading to an amelioration of the arthritis in SKG mice. C968 inhibited the cell cycle of RA-FLSs, which slowed the progression of arthritis in SKG mice.
HK2, GAPDH, ENO, and PKM2 can migrate into the nucleus, and act as a transcriptional or translational factor. LDH-A is associated with acetyl-CoA consumption in the cytosol and mitochondria. An increase in lactate production in the cytosol leads to a lack of acetyl-CoA in the mitochondria, resulting in a decrease in histone acetylation. HK2: hexokinase 2, GAPDH: glyceraldehide-3-phosphate dehydrogenase, ENO: enolase, PKM2: pyruvate kinase muscle 2, LDHA: lactate dehydrogenase-A.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Koppenol WH, Bounds PL, Dang CV, et al. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337.
- Palsson-McDermott EM, O'Neill LAJ. The Warburg effect then and now: from cancer to inflammatory diseases. Bioessays. 2013;35:965–973.
- Ghesquière B, Wong BW, Kuchnio A, et al. Metabolism of stromal and immune cells in health and disease. Nature. 2014;511:167–176.
- Millet P, Vachharajani V, McPhail L, et al. GAPDH binding to TNF-αmRNA contributes to posttranscriptional repression in monocytes: a novel mechanism of communication between inflammation and metabolism. JI. 2016;196:2541–2551.
- Bustamante MF, Garcia-Carbonell R, Whisenant KD, et al. Fibroblast-like synoviocyte metabolism in the pathogenesis of rheumatoid arthritis. Arthritis Res Ther. 2017;19:110.
- Young SP, Kapoor SR, Viant MR, et al. The impact of inflammation on metabolomic profiles in patients with arthritis. Arthritis Rheum. 2013;65:2015–2023.
- Lauridsen MB, Bliddal H, Christensen R, et al. 1H NMR spectroscopy-based interventional metabolic phenotyping: a cohort study of rheumatoid arthritis patients. J Proteome Res. 2010;9:4545–4553.
- Madsen RK, Lundstedt T, Gabrielsson J, et al. Diagnostic properties of metabolic perturbations in rheumatoid arthritis. Arthritis Res Ther. 2011;13:R19.
- Yang XY, Zheng KD, Lin K, et al. Energy metabolism disorder as a contributing factor of rheumatoid arthritis: a comparative proteomic and metabolomic study. PLoS One. 2015;10:e0132695.
- Zabek A, Swierkot J, Malak A, et al. Application of (1)H NMR-based serum metabolomic studies for monitoring female patients with rheumatoid arthritis. J Pharm Biomed Anal. 2016;117:544–550.
- Guo H, Niu X, Gu Y, et al. Differential amino acid, carbohydrate and lipid metabolism perpetuations involved in a subtype of rheumatoid arthritis with chinese medicine cold pattern. IJMS. 2016;17:e1757.
- Li J, Che N, Xu L, et al. LC-MS-based serum metabolomics reveals a distinctive signature in patients with rheumatoid arthritis. Clin Rheumatol. 2018;37:1493–1502.
- Jin L, Alesi GN, Kang S, et al. Glutaminolysis as a target for cancer therapy. Oncogene. 2016;35:3619–3625.
- Cooper EH, Barkhan P, Hale AJ, et al. Observations on the proliferation of human leucocytes cultured with phytohaemagglutinin. Br J Haematol. 1963;9:101–111.
- Guppy M, Greiner E, Brand K, et al. The role of the Crabtree effect and an endogenous fuel in the energy metabolism of resting and proliferating thymocytes. Eur J Biochem. 1993;212:95–99.
- Frauwirth KA, Riley JL, Harris MH, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16:769–777.
- Buttgereit F, Burmester GR, Brand MD, et al. Bioenergetics of immune functions: fundamental and therapeutic aspects. Immunol Today. 2000;21:192–199.
- Krauss S, Brand MD, Buttgereit F, et al. Signaling takes a breath–new quantitative perspectives on bioenergetics and signal transduction. Immunity. 2001;15:497–502.
- Wang R, Dillon CP, Shi LZ, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882.
- Chang C-H, Curtis JD, Maggi LB, et al. Posttranscriptional control of t cell effector function by aerobic glycolysis. Cell. 2013;153:1239–1251.
- Kolev M, Dimeloe S, Le Friec G, et al. Complement regulates nutrient influx and metabolic reprogramming during Th1 cell responses. Immunity. 2015;42:1033–1047.
- Shi LZ, Wang R, Huang G, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208:1367–1376.
- Araujo L, Khim P, Mkhikian H, et al. Glycolysis and glutaminolysis cooperatively control T cell function by limiting metabolite supply to N-glycosylation. Elife. 2017;6:e21330.
- Berod L, Friedrich C, Nandan A, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med. 2014;20:1327–1333.
- Michalek RD, Gerriets VA, Jacobs SR, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186:3299–3303.
- Sundrud MS, Koralov SB, Feuerer M, et al. Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science. 2009;324:1334–1338.
- Everts B, Amiel E, Huang SC-C, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKɛ supports the anabolic demands of dendritic cell activation. Nat Immunol. 2014;15:323–332.
- Sim WJ, Ahl PJ, Connolly JE, et al. Metabolism is central to tolerogenic dendritic cell function. Mediators Mediators Inflamm. 2016;2016:1.
- Ferreira GB, Kleijwegt FS, Waelkens E, et al. Differential protein pathways in 1,25-dihydroxyvitamin d(3) and dexamethasone modulated tolerogenic human dendritic cells. J Proteome Res. 2012;11:941–971.
- Rhoads JP, Major AS, Rathmell JC, et al. Fine tuning of immunometabolism for the treatment of rheumatic diseases. Nat Rev Rheumatol. 2017;13:313–320.
- Seki SM, Gaultier A. Exploring non-metabolic functions of glycolytic enzymes in immunity. Front Immunol. 2017;8:1549.
- Kotake S, Udagawa N, Takahashi N, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. 1999;103:1345–1352.
- Sato K, Suematsu A, Okamoto K, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203:2673–2682
- Zhu J, Yamane H, Paul W. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. 2010;28:445–489.
- Wang W, Shao S, Jiao Z, et al. The Th17/Treg imbalance and cytokine environment in peripheral blood of patients with rheumatoid arthritis. Rheumatol Int. 2012;32:887–893.
- Yang Z, Fujii H, Mohan SV, et al. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J Exp Med. 2013;210:2119–2134.
- Sena LA, Li S, Jairaman A, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. 2013;38:225–236.
- Yang Z, Shen Y, Oishi H, et al. Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci Transl Med. 2016;8:331ra38.
- Sun L, Fu J, Zhou Y, et al. Metabolism controls the balance of Th17/T-regulatory cells. Front Immunol. 2017;8:1632.
- Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. 2013;9:24–33.
- Takahashi S, Saegusa J, Sendo S, et al. Glutaminase 1 plays a key role in the cell growth of fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res Ther. 2017;19:76.
- Henderson B, Bitensky L, Chayen J, et al. Glycolytic activity in human synovial lining cells in rheumatoid arthritis. Ann Rheum Dis. 1979;38:63–67.
- Ahn JK, Kim S, Hwang J, et al. Metabolomic elucidation of the effects of curcumin on fibroblast-like synoviocytes in rheumatoid arthritis. PLoS One. 2015;10:e0145539.
- Ahn JK, Kim S, Hwang J, et al. GC/TOF-MS-based metabolomic profiling in cultured fibroblast-like synoviocytes from rheumatoid arthritis. Joint Bone Spine. 2016;83:707–713.
- Okano T, Saegusa J, Nishimura K, et al. 3-bromopyruvate ameliorate autoimmune arthritis by modulating Th17/Treg cell differentiation and suppressing dendritic cell activation. Sci Rep. 2017;10:42412.
- Panneton V, Bagherzadeh Yazdchi S, Witalis M, et al. ICOS signaling controls induction and maintenance of collagen-induced arthritis. J Immunol. 2018;200:3067–3076.
- Bian L, Josefsson E, Jonsson I-M, et al. Dichloroacetate alleviates development of collagen II-induced arthritis in female DBA/1 mice. Arthritis Res Ther. 2009;11:R132.
- Garcia-Carbonell R, Divakaruni AS, Lodi A, et al. Critical role of glucose metabolism in rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Rheumatol. 2016;68:1614–1626.
- Fox CJ, Hammerman PS, Thompson CB, et al. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol. 2005;5:844–852.
- Lai R, Xian D, Xiong X, et al. Proanthocyanidins: novel treatment for psoriasis that reduces oxidative stress and modulates Th17 and Treg cells. Redox Rep. 2018;23:130–135.
- Essig K, Hu D, Guimaraes JC, et al. Roquin suppresses the PI3K-mTOR signaling pathway to inhibit T helper cell differentiation and conversion of Treg to Tfr Cells. Immunity. 2017;47:1067–1082;e12.
- Binger KJ, et al. Immunometabolic regulation of interleukin-17-producing T helper cells: uncoupling new targets for autoimmunity. Front Immunol. 2017;8:311.
- Ma H, Botstein D. Effects of null mutations in the hexokinase genes of Saccharomyces cerevisiae on catabolite repression. Mol Cell Biol. 1986;6:4046–4052.
- Moreno F, Herrero P. The hexokinase 2-dependent glucose signal transduction pathway of Saccharomyces cerevisiae. FEMS Microbiol Rev. 2002;26:83–90.
- Ahuatzi D, Herrero P, de la Cera T, et al. The glucose-regulated nuclear localization of hexokinase 2 in Saccharomyces cerevisiae is Mig1-dependent. J Biol Chem. 2004;279:14440–14446.
- Saad S, Peter M, Dechant R, et al. In scarcity and abundance: metabolic signals regulating cell growth. Physiology (Bethesda)). 2013;28:298–309.
- Lv L, Xu Y-P, Zhao D, et al. Mitogenic and oncogenic stimulation of K433 acetylation promotes PKM2 protein kinase activity and nuclear localization. Mol Cell. 2013;52:340–352.
- Shirai T, Nazarewicz RR, Wallis BB, et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J Exp Med. 2016;213:337–354.
- Luo W, Hu H, Chang R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–744.
- Gao X, Wang H, Yang JJ, et al. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell. 2012;45:598–609.
- Demaria M, Poli V. PKM2, STAT3 and HIF-1α: The Warburg's vicious circle. JAKSTAT. 2012;1:194–196.
- Feo S, Arcuri D, Piddini E, et al. ENO1 gene product binds to the c-myc promoter and acts as a transcriptional repressor: relationship with Myc promoter-binding protein 1 (MBP-1). FEBS Lett. 2000;473:47–52.
- Ray R, Miller DM. Cloning and characterization of a human c-myc promoter-binding protein. Mol Cell Biol. 1991;11:2154–2161.
- De Rosa V, Galgani M, Porcellini A, et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat Immunol. 2015;16:1174–1184.
- Du J, Huang C, Zhou B, et al. Isoform-specific inhibition of ROR α-mediated transcriptional activation by human FOXP3. J Immunol. 2008;180:4785–4792.
- Peng M, Yin N, Chhangawala S, et al. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science. 2016;354:481–484.
- Pioli PA, Hamilton BJ, Connolly JE, et al. Lactate dehydrogenase is an AU-rich element-binding protein that directly interacts with AUF1. J Biol Chem. 2002;277:35738–35745.