
Abstract
T cells recognize antigen peptides displayed by HLA molecules and specifically eliminate their target cells. Identification of responsible antigens as well as understanding the mechanism by which antigens are produced inside cells are equally crucial for cancer immunology. In this review, we introduce proteogenomics and its applications in cancer antigen research, which leverages mass spectrometry and next-generation sequencing. The approach comprehensively captures immunopeptidome displayed by HLA, revealing new classes of antigens, such as mutation-derived neoantigens, spliced peptides, and non-coding region derived peptides. These antigens may serve as therapeutic targets or biomarkers. Thus, proteogenomics is a promising approach for cancer antigen research and contributes to immunotherapy development.
Keywords:
1. Introduction
Cytotoxic T lymphocytes (CTL) or CD8+ T cells recognize peptide-HLA class I complexes (pHLA I) on the surface. The antigen presented by HLA class I is a hallmark of CTL discrimination between self and non-self, such as virus-infected or cancer cells. Immune checkpoint inhibitors (ICI) have brought about benefits for patients with a wide range of cancer types, demonstrating that immune system is capable of recognizing and eliminating tumors. The immunotherapy using antibodies against PD-1, PD-L1, and CTLA-4 primarily affects patient T-cell functions and restores their killing activity against cancer cells. However, despite of its clinical success, objective response rates are not yet satisfactory (20–30% for many types of cancer), calling for additional targets or prediction biomarkers [Citation1,Citation2]. One major concern is a paucity of knowledge about antigens responsible for T-cell cancer discrimination. Mutation-derived neoantigens likely play a principal role; however, therapeutic effects are not always limited to patients with high-mutation burden cancer, implying a diverse range of cancer antigen source presented by HLA molecules [Citation3]. Because any type of cancer aberration recognized by CTLs is essential for therapeutic application, comprehensive analysis of the cancer antigens as well as development of natural T-cell epitope prediction are in great demand. HLA ligandome analysis makes use of biochemical isolation of HLA-bound peptides followed by mass spectrometry sequencing, directly revealing an immunopeptidome of cancer cells. Here, we review the mass spectrometry use for cancer antigen research as well as advances in proteogenomic approaches to detect new classes of antigens.
2. Mass spectrometry contribution to understand HLA class I antigen processing
The antigen processing begins in the cytoplasm where endogenous proteins are digested by the proteasomes. The fragmented polypeptides are transported into the endoplasmic reticulum (ER) through the transporter associated with antigen processing (TAP). In the ER, the peptide precursors are trimmed by the ER-resident aminopeptidase associated with antigen processing (ERAP1 or ERAAP) and optimized for HLA class I binding. Peptide loading complex (PLC) that comprises tapasin, ERp53, calreticulin, and TAP helps form stable pHLA I most likely by peptide exchange. Lack of any of the machinery influences the presentation mechanism, thereby alters pHLA I surface repertoire. Thus, pHLA I on display samples a wide range of ‘gene-chips’ of the cell according to the elaborated, but complicated, antigen-processing pathway [Citation4,Citation5].
Current prevalent algorithms (e.g., NetMHC) predicting natural peptides are trained using datasets captured by biochemical assays using mass spectrometry [Citation6]. Advances in the sensitivity have contributed to build more reliable algorithms. Among the factors that predict natural presentation, presence of HLA binding anchors within a sequence is arguably the most prominent feature, which defines the binding affinity between a peptide and HLA molecule. However, affinity prediction alone often ends up with a heap of false positives. There is no definitive threshold to judge presentation, and a lower threshold to predict more peptides concomitantly increases irrelevant hit rates. One sensible solution is to filter out false positives according to their mRNA expression levels, because abundant expression is positively correlated with peptide presentation [Citation7]. Another potential solution is to trace the footprints of natural peptide sequences to decipher preferences in the antigen processing pathway. Abelin and colleagues have recently demonstrated that not only peptide sequences themselves but also surrounding upstream and downstream sequences are biased in natural peptides displayed by several cell lines including cancer [Citation8]. Our group has also showed that the protein sequences following proline are unfavorable for HLA presentation owing to the antigen processing mechanism inside the ER () [Citation9]. Prediction of the peptide selection through the antigen processing pathway is yet to be perfect; nevertheless, the integration of natural HLA ligandome datasets across different HLA/cell types would accelerate its development.
Figure 1. Proline inhibits HLA class I presentation of following epitopes. The heat map shows the frequency profile of Upstream and downstream residues surrounding natural HLA class I epitopes (log2[FoldChange]). Data were calculated using more than 2000 natural ligands captured from cancer cells with multiple HLA class I types. Depletion of proline at U1-3 indicates that peptide sequences following the proline residue are barely presented by HLA class I (adopted from Ref. [Citation9]).
![Figure 1. Proline inhibits HLA class I presentation of following epitopes. The heat map shows the frequency profile of Upstream and downstream residues surrounding natural HLA class I epitopes (log2[FoldChange]). Data were calculated using more than 2000 natural ligands captured from cancer cells with multiple HLA class I types. Depletion of proline at U1-3 indicates that peptide sequences following the proline residue are barely presented by HLA class I (adopted from Ref. [Citation9]).](/cms/asset/3fd36fcb-6afd-4e77-81b2-8d1fb427482f/timm_a_1640500_f0001_c.jpg)
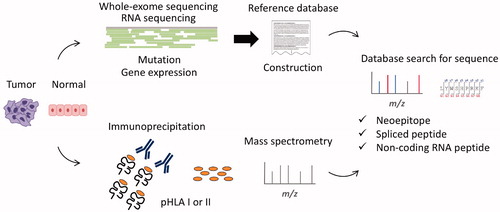
3. Proteogenomics HLA ligandome analysis
Technological advances in mass spectrometry, along with development of orbitrap analysis, have enhanced its resolution and consequently enabled to sequence thousands of natural peptides displayed by HLA class I of cells. Although de novo sequencing is available (e.g., PEAKS), a reference protein database is indispensable for MS spectral matching and commonly used to maximize the accuracy and number of peptides. However, such generalized protein databases (e.g., UniProt) do not contain information unique to individuals (e.g., cancer somatic mutation), thereby struggles to yield corresponding peptide types (e.g., neopeitope). To address this issue, proteogenomics combines conventional proteomics with genomics using next-generation sequencing (NGS) (). For example, somatic mutations detected by whole-exome sequencing (WES) are converted to amino-acid sequences, and incorporated to the reference database for neoantigen detection. RNA-seq-based gene expression analysis also contributes to leave out unnecessary entries and optimize the database. Gene mutation is not the sole target of the proteogenomic approach: as described below, proteogenomics HLA ligandome analysis captures a variety of unique peptides, which otherwise had been difficult to be shown. Recent technological advances in both mass spectrometry and NGS availability expanded a range of antigen detection, and enable to sequence the unprecedented numbers of HLA ligands including the ones that are not mapped as conventional proteins [Citation10–12]. On the other hand, one major remaining issue is that, even though the sensitivity has been significantly improved in the decade, yet the proteomic analysis requires a certain amount of samples, compared with the genomics part, which hinders its application to tiny biopsy samples.
Figure 2. Proteogenomics HLA ligandome analysis workflow. Peptide-HLA class I or II complexes (pHLA I or II) are captured from tumor/normal cells, then only peptides bound to HLA are analyzed using mass spectrometry. Meanwhile, genomics data from WES (e.g., mutation) or RNA-seq (e.g., expression) are used to make its reference database of interest. The database is searched for each MS/MS spectrum to sequence the corresponding peptide sequence. Protegenomics enables identification of somatic mutation-derived neoantigens, spliced peptides, and non-coding region derived peptides.
4. Neoantigen
Current evidences show that cancer-specific T cells recognize neoantigens (or neoepitope) that arise from somatic gene mutation. ICIs are often effective on cancer with high mutation burden, suggesting that non-synonymous mutation creates neoepitopes that harbor an amino-acid substitution for HLA presentation [Citation13]. Because neoepitopes are not presented by normal counterpart cells, or thymic epithelial cells, T cells recognizing the neoepitopes are not subject to central tolerance in the thymus. Hence, it is conceivable that neoantigens containing neoepitopes are in charge of eliciting strong and specific host T cell responses against cancer. Meanwhile, detecting neoepitopes is a daunting task. Neoepitopes can be predictable from given protein sequences using prediction algorisms (e.g., NetMHC) along with mutation data from WES analysis; however, it is yet uncertain whether each candidate epitope is naturally presented by HLA molecules of cancer cells, unless recognition of the primary cancer cells is demonstrated using the specific T cells [Citation14]. There are also factors impeding the prediction. For example, loss of heterozygosity (LOH) is observed in the HLA locus potentially responsible for neoepitope presentation in early stage lung cancer [Citation15]. The antigen processing machinery is often diminished in a variety of cancer as well, which could alter antigen presentation and result in loss of cancer antigen presentation [Citation16]. In addition, there are further issues in predicting HLA class II neoepitopes owing to their promiscuous binding motifs and variation in peptide lengths [Citation17]. Besides HLA class I, recent studies suggest a critical role of HLA class II neoepitopes in patient anti-cancer T-cell responses [Citation18,Citation19].
Proteogenomic approaches can be used to address these obstacles, allowing to detect naturally presented HLA class I and II neoepitopes. Because neoepitopes on display are, by and large, far less than those candidates predicted solely using in silico algorithms, direct detection using proteogenomics significantly benefits the neoantigen research [Citation20]. In our own study, we identified a naturally presented HLA-A24 neoepitope (AKF9), which is a 9-mer peptide that arouse from a passenger mutation (c.258C > G) of the ubiquitously expressed AP2S1 gene [Citation21]. The AKF9 elicited CTL responses that exhibited considerably high and specific cytotoxic activity against the colon cancer cells carrying the mutation.
However, the prevalence of such an immunogenic neoantigen still remains unclear. One can argue that only about 1% of whole exomic non-synonymous mutations are potentially recognized by tumor-infiltrating CTLs as neoepitopes [Citation22]. Although proteogenomics can be applied to primary cancer tissues, yet the successful reports are limited to date [Citation23]. Further studies would clarify the HLA neoepitope repertoire in clinical samples as well as their hierarchy of immunogenicity to elicit patient T-cell responses.
5. Spliced peptide
Proteogenomic approach shows its real worth in search of unconventional of antigens, which otherwise had been difficult to be proved. It has been known that the proteasomes not only digest but also splice two peptide fragments together, creating peptide splice variants [Citation24]. This particular event is intriguing because splicing may increase HLA ligand production. Liepe et al. have reported that in fact the HLA class I ligands produced by peptide splicing exist, and to our surprise, they consist of about one-third of the whole ligandome in multiple cell types [Citation25]. The result implies that new peptides harboring appropriate HLA-binding anchors can be created during antigen processing pathway. Because a single spliced peptide can arise from non-consecutive sequences mapped in the genome even across multiple genes, this could be an intrinsic mechanism to expand a diversity of HLA ligands on display [Citation26]. Although the spliced peptides were initially identified as cancer antigens, their contribution to cancer immunology yet remains elusive [Citation27]. We hypothesize that such a large pool of peptides likely give rise to neoepitopes.
6. Non-coding RNA antigen
While conventional proteomics focuses on protein-coding genes, which account for only a few percent of the genome, about three-quarters of the human genome is indeed transcribed [Citation28]. The majority of the transcripts are non-coding or unannotated RNAs. Even though their ‘potential’ open reading frames are too short to create functional proteins, quite a few long non-coding RNAs possess both 5’ cap structures and poly-A tails, allowing ribosome binding [Citation29]. It has long been proposed that those non-coding RNAs are a potential source of HLA ligands [Citation30]. Recently, by means of proteogenomic approaches, Laumont et al. and our group have independently proved the presence of cancer-specific MHC class I antigens derived from non-coding region, in mouse and human, respectively. Laumont et al. have shown that the immunization of the non-coding RNA antigens elicited CTL responses thereby contributed to overall survival in vivo mouse cancer models [Citation31]. Moreover, host anti-cancer CTL responses were biased toward the non-coding derived peptides than conventional ones. In our study, an antigen derived from a long non-coding RNA was presented by HLA class I of primary cancer tissues, inducing CTL responses specific to the cancer cells expressing the gene (data not shown). These studies demonstrate that translation followed by MHC presentation could happen in clinical settings, and allegedly non-coding RNAs give rise to cancer antigens that elicit host CTL responses. In contrast to neoantigens that arise from passenger mutations, non-coding RNA antigens are not necessarily patient specific but indeed detected across individuals. Therefore, they may serve as attractive targets of vaccination or adoptive T-cell transfer therapies.
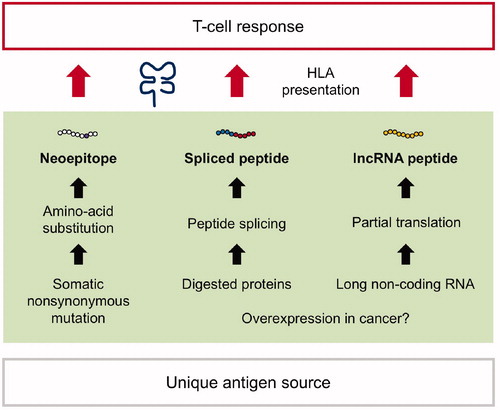
Currently, little is known about the mechanisms underlying its cancer specificity. As a whole, immunogenicity to elicit anti-cancer host CTL response is attributed to cancer-specific antigen presentation. Are the responsible non-coding genes simply overexpressed in cancer in context of oncogenesis? Or, is there a cancer-specific mechanism allowing the unconventional translation from non-coding regions? The clarification would highlight the new class of cancer antigens and benefit its therapeutic application. The generation and HLA presentation of three classes of unique antigens mentioned in this review (neoantigens, spliced peptides, and non-coding RNA peptides) are summarized in .
Figure 3. Discovery of unique classes of cancer antigens. (Left) Non-synonymous mutations (missense or frameshift mutations) give rise to unique amino-acid substitutions and can be presented by HLA as neoepitopes. Because somatic mutation is a cancer specific event, the neoepitopes are often immunogenic to induce host T-cell anti-cancer responses. (Middle) The proteasomes can ligate two excised peptide fragments and create a single peptide (peptide splicing). The post-translational event provides spliced peptides, which can be presented by HLA and serve as T-cell targets. (Right) Non-coding genes do not harbor evident open-reading frames encoding functional proteins. However, partial translation can occur, yielding HLA-bound peptides (long non-coding RNA peptide, lncRNA peptide). Both spliced peptides and lncRNA peptides can elicit anti-cancer responses as well; however, the underlying mechanisms remain unclear, therefore need to be investigated.
7. Concluding remark
Recent advances in proteogenomics HLA ligandome analysis have significantly broaden a range of cancer antigen research. The approach provides the most reliable way to look into the natural HLA peptide repertoire, directly proving the presentation of otherwise conceptual antigens. Discovery of diverse classes of cancer antigens suggests that T cells are capable of sensing a variety of cancer aberration. Presumably, the antigen diversity implies the possibility that patient T cells react not only a single but also multiple classes of antigens, and the preference in host immune surveillance could differ in individuals. Thus, the hierarchy in patient T cell responses should be further investigated using clinical samples, which would ultimately lead to development of biomarkers or precision medicine for cancer immunotherapy. In addition, because there are still a plenty of room for its application, the approach also contributes to any of immunology research as well as cancer immunology.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–330.
- Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19:133.
- Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2017;377:2500–2501.
- Rock KL, Reits E, Neefjes J. Present yourself! By MHC Class I and MHC Class II molecules. Trends Immunol. 2016;37:724–737.
- Shastri N, Schwab S, Serwold T. Producing nature's gene-chips: the generation of peptides for display by MHC class I molecules. Annu Rev Immunol. 2002;20:463–493.
- Lundegaard C, Lamberth K, Harndahl M, et al. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8-11. Nucleic Acids Res. 2008;36:W509–W512.
- Bassani-Sternberg M, Pletscher-Frankild S, Jensen LJ, et al. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol Cell Proteomics. 2015;14:658–673.
- Abelin JG, Keskin DB, Sarkizova S, et al. Mass spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity. 2017;46:315–326.
- Hongo A, Kanaseki T, Tokita S, et al. Upstream position of proline defines peptide-HLA Class I repertoire formation and CD8(+) T cell responses. J Immunol. 2019;202:2849–2855.
- Creech AL, Ting YS, Goulding SP, et al. The role of mass spectrometry and proteogenomics in the advancement of HLA epitope prediction. Proteomics. 2018;18:1700259.
- Freudenmann LK, Marcu A, Stevanovic S. Mapping the tumour human leukocyte antigen (HLA) ligandome by mass spectrometry. Immunology. 2018;154:331–345.
- Laumont CM, Perreault C. Exploiting non-canonical translation to identify new targets for T cell-based cancer immunotherapy. Cell Mol Life Sci. 2018;75:607–621.
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74.
- Vitiello A, Zanetti M. Neoantigen prediction and the need for validation. Nat Biotechnol. 2017;35:815–817.
- McGranahan N, Rosenthal R, Hiley CT, et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell. 2017;171:1259.
- Shionoya Y, Kanaseki T, Miyamoto S, et al. Loss of tapasin in human lung and colon cancer cells and escape from tumor-associated antigen-specific CTL recognition. Oncoimmunology. 2017;6:e1274476.
- EDITORIAL. The problem with neoantigen prediction. Nat Biotechnol. 2017;35:97.
- Ott PA, Hu Z, Keskin DB, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217.
- Marty R, Thompson WK, Salem RM, et al. Evolutionary pressure against MHC Class II binding cancer mutations. Cell. 2018;175:416–428.e413.
- Yadav M, Jhunjhunwala S, Phung QT, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515:572–576.
- Kochin V, Kanaseki T, Tokita S, et al. HLA-A24 ligandome analysis of colon and lung cancer cells identifies a novel cancer-testis antigen and a neoantigen that elicits specific and strong CTL responses. Oncoimmunology. 2017;6:e1293214.
- Tran E, Ahmadzadeh M, Lu Y-C, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015;350:1387–1390.
- Bassani-Sternberg M, Bräunlein E, Klar R, et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat Commun. 2016;7:13404.
- Vigneron N, Ferrari V, Stroobant V, et al. Peptide splicing by the proteasome. J Biol Chem. 2017;292:21170–21179.
- Liepe J, Marino F, Sidney J, et al. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science. 2016;354:354–358.
- Faridi P, Li C, Ramarathinam SH, et al. A subset of HLA-I peptides are not genomically templated: evidence for cis- and trans-spliced peptide ligands. Sci Immunol. 2018;3:eaar3947.
- Hanada K, Yewdell JW, Yang JC. Immune recognition of a human renal cancer antigen through post-translational protein splicing. Nature. 2004;427:252–256.
- Djebali S, Davis CA, Merkel A, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108.
- Ingolia NT. Ribosome footprint profiling of translation throughout the genome. Cell. 2016;165:22–33.
- Boon T, Van Pel A. T cell-recognized antigenic peptides derived from the cellular genome are not protein degradation products but can be generated directly by transcription and translation of short subgenic regions. A hypothesis. Immunogenetics. 1989;29:75–79.
- Laumont CM, Vincent K, Hesnard L, et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci Transl Med. 2018;10:eaau5516.