
Abstract
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease which affects the majority of organs and systems. Traditional therapies do not lead to complete remission of disease but only relieve symptoms and inflammation. B cells are the most important effector cell types in the pathogenesis of SLE. Therefore, therapies targeting B cells and their related cytokines are a very important milestone for SLE treatment. Several biologics that modulate B cells, either depleting B cells or blocking B cell functions, have been developed and evaluated in clinical trials. Belimumab, a fully humanized monoclonal antibody that specifically binds B cells activating factor (BAFF), was the first of these agents approved for SLE treatment. In this review, we explore the currently available evidence in B cell targeted therapies in SLE including agents that target B cell surface antigens (CD19, CD20, CD22), B cell survival factors (BAFF and a proliferation-inducing ligand, APRIL), cytokines (interleukin-1 and type 1 interferons) and co-stimulatory molecules (CD40 ligand). We highlighted the mechanisms of action and the individual characteristics of these biologics, and present an update on the clinical trials that have evaluated their efficacy and safety. Finally, we describe some of the emerging and promising therapies for SLE treatment.
1. Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease that can affect the majority of organs and systems. The clinical presentations include a great diversity of symptoms ranging from mild to severe disease, with periods of remissions and flares. SLE has an unpredictable prognosis, depending mainly on severity of the major organ involvement, and an increased mortality with an estimated standardized mortality rate of 2.4–5.9 [Citation1,Citation2].
Traditional therapies including corticosteroids, hydroxychloroquine, azathioprine, cyclophosphamide, mycophenolate mofetil (MMF) or tacrolimus have been proved effective for SLE patients reducing organ damage and mortality. However, these agents have broadly immunosuppressive mechanisms of actions causing mild-to moderate adverse effects [Citation3,Citation4]. Despite many advances have been made in the treatment of SLE resulting in an increase of short- and medium-term survival [Citation5], the rate of premature deaths, long-term prognosis and damage accrual still remains poor especially in patients with major organ involvement.
Hence, prevention and minimization of damage accrual and pursuing a stable disease controls are the main target in SLE, the utility of traditional agents is limited due to the serious adverse effects, and it is necessary to search for more specific and less toxic therapies with at least similar effectivity.
The precise mechanisms underlying disease pathogenesis in SLE are not fully understood, though it is well established that defective immune regulatory mechanisms, such as the clearance of apoptotic cells and immune complexes, are important contributors to SLE development [Citation6]. The loss of immune tolerance leads to activation of autoreactive T cells and B cells. These cells proliferate and differentiate into harmful pathogenic T cells and plasma B cells producing pathogenic autoantibodies [Citation7,Citation8]. Recent insights into the pathogenesis of autoimmune diseases led to the development of a novel class of agents which targets specific dysregulated components of the immune system. Targeted immunomodulatory therapies in rheumatic diseases were first introduced in the late 1990s [Citation9,Citation10]. Autoreactive B cells are one of the effector cell types responsible for the perpetuation of the inflammation responses. Therefore, therapies targeting B cells and their related cytokines are a very important milestone for SLE treatment [Citation11].
In 2011, the United States Food and Drug Administration (FDA) and European Medicine Agency (EMA) approved belimumab, the first medication to be licensed exclusively for the treatment of SLE since 1957 [Citation12]. Belimumab is a fully humanized monoclonal antibody designed to specifically target the B cells activating factor (BAFF), preventing BAFF interaction with BAFF-related receptors, hence reducing the numbers of peripheral naïve, transitional and activated B cells [Citation13].
In the last years, several targeted therapies have been developed and evaluated in clinical trials. However, the heterogeneity in the clinical manifestation of SLE had a great impact in the success or failure of these agents in clinical trials. To standardize the outcome of drug efficacy in SLE clinical trials, several disease activity indexes (DAI) have been used, according to the FDA recommendation. Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) includes 24 clinical and laboratory variables of 9 organ systems that are weighted by the type of manifestation but not by severity. Safety of Estrogens in Lupus National Assessment study-SLEDAI (SELENA-SLEDAI) is a modified version of SLEDAI with addition scoring of some descriptors such as rash, mucosal ulcers and alopecia. The British Isles Lupus Assessment Group (BILAG) index is an organ-based transitional activity instrument which provides disease activity scorings across eight organ systems on an ordinal scale (A to E) based on the physician’s intention-to-treat premise. BILAG-Based Composite Lupus Assessment (BICLA) responder is referred as patients who meet BILAG improvement in A and B scores, no more than one new BILAG B score, no worsening of total SLEDAI score from baseline, less than 10% deterioration and no initiation of non-protocol treatment. SLE Responder Index (SRI) is a composite outcome including a modification of SELENA-SLEDAI, BILAG and a 3-cm visual analog scale of physician-rated disease activity (PGA). Cutaneous Lupus Erythematosus Disease Area and Severity Index (CLASI) is a tool for assessing disease activity and damage in cutaneous lupus erythematosus (CLE) [Citation14].
This review describes the current available evidence in B cell target therapies in SLE. Furthermore, we describe the emerging targeted therapies with great potential in SLE management.
2. Roles of B cells in SLE
In physiological development, B cells are capable to differentiate into antibody-secreting plasmablasts and plasma cells. B cell development occurs in in the fetal liver and adult bone marrow. Pre-B cells arise from progenitor (pro-B) cells and differentiate into immature B cells. Early B cell development is characterized by a molecular progress to rearrange the heavy and light chains of their immunoglobulin (Ig) genes. This process is supported by stromal cell-deriver interleukin (IL)-7 [Citation15]. When the rearrangement is completed, B cells express a unique B-cell receptor (BCR) required for further development and survival [Citation16].
Immature B cells exiting from bone marrow enter a transitional phase during which further maturation events occur to produce mature cells. B cells migrate to secondary lymphoid organs (spleen or lymph nodes) where may encounter antigen through interact with dendritic cells or macrophages. B cells can enter in a germinal center or differentiate into a short-lived plasma cell. Within the germinal center, B cells undergo clonal expansion, class switch recombination or somatic hypermutations resulting in antibody-secreting cells, including plasmablasts and plasma cells, and memory cells [Citation17]. B cells can also be divided in marginal zone B cell subsets found in the marginal zone of the spleen, or follicular zone B cell subsets circulating in the periphery. Memory cells generated in the germinal center persist and differentiate into plasma cells in a secondary immune response to provide rapid antibody production [Citation18].
In SLE, plasmablasts and plasma cells produce a wide spectrum of autoantibodies which are considered highly relevant for the disease pathogenesis such antibodies against double stranded (ds) or single (ss) stranded DNA, RNA containing/binding nuclear antigens including Smith (Sm) antigen, ribonucleoprotein, the Ro and La antigens, ribosomal P protein or phospholipids. Different types of autoantibodies are associated with different clinical presentations in SLE. Anti-dsDNA antibodies are one of the most common autoantibodies in SLE patients and their serum levels associated with renal disease in SLE [Citation19].
Besides the production of autoantibodies, B cells may also function as antigen-presenting cells (APCs), assisting in the activation of autoreactive T cells [Citation20]. The antibody-independent role of B cells was highlighted by Chan et al. [Citation21]. They created a mutant mouse (MRL/lpr) with B cells that only expressed a transgene encoding surface Ig but which did not permit the secretion of antibodies. These mice developed nephritis with cellular infiltration within the kidney and spontaneous T cell activation indicating that B cells themselves, without soluble antibody production, exert pathogenic role. Furthermore, autoantigen primed B cells can activate autoreactive T cells in vivo [Citation22]. Therefore, functional B cells independent of autoantibody production, are essential for disease expression, either contributing directly to local inflammation or by serving as APCs for antigen-specific autoreactive T cells.
Further, B cells have a role as cytokine producers which contribute to modulate the magnitude of the immune response [Citation23]. The profile of human B cell cytokine secretion depends on the stimuli they encounter through the BCR and CD40. Sequential stimulation of BCR and CD40 in B cells leads to B cell proliferation and secretion of pro-inflammatory cytokines such as tumor necrosis factor (TNF)α, lymphotoxin and IL-6 which may act as autocrine growth and differentiation factors, as well as, serve to amplify the ongoing immune response. In contrast, CD40 stimulation alone induces a significant production of IL-10 which suppressed “inappropriate” immune response. Therefore, cytokine secretion by B cells require additional signaling beyond activation which can be provided by the immune microenvironment and specific stages of B cell differentiation [Citation24]. Many cytokine pathways have been involved in the pathogenesis of SLE. Cytokines produce during immune activation have pleiotropic effects and can contribute positively or negatively to the expression or function of other cytokines. Owing the heterogeneity of SLE manifestations, it is probable that the contribution of B-cell derived cytokines differs between individual patients [Citation25].
3. B cells as therapeutic target in SLE
SLE is characterized by polyclonal B-cell over-reactivity which might be related to intrinsic polyclonal B cell activation with disturbed activation thresholds, and ineffective negative selection, lack of immunoregulatory functions, overactive inflammatory environment or disturbed cytokine production by non-B immune cells. These mechanisms are not mutually exclusive and may operate to varying extents and at different times in SLE [Citation26–28].
Therapies to selective deplete B cells or reduce B cell-related cytokines became attractive therapeutic approaches in SLE. However, an important raised issue is the long-term safety of B cell depleting agents, and it has been suggested that inhibition of B cell rather than depletion might be a preferred approach.
Many agents have been developed, some of them target specific surface antigens expressed across different B-cell subsets including CD19, CD20, CD22 [Citation29–31]. Other agents are directed against the BAFF/a proliferation-inducing ligand (APRIL) system, cytokines such as IL-6 and its receptor (IL-6R), interferon (IFN) or CD40-CD40 ligand co-stimulatory system [Citation32–39] ().
Table 1. Target molecules for B cell therapies.
3.1. Targeting B cell-surface antigens
3.1.1. Anti-CD19 targeting therapies
CD19 is a transmembrane glycoprotein of the Ig superfamily which is expressed throughout the whole B cells development from the pre-B cells to the plasma cells [Citation40]. CD19 acts as an adaptor protein to recruit cytoplasmic signaling proteins to the membrane. It also works within the CD19/CD21 complex, as a positive co-receptor of BCR enhancing BCR signaling in response to T cell-dependent complement-tagged antigens [Citation41,Citation42]. In contrast, a negative co-receptor for BCR, Fcγ receptor (FcγR) IIb, the only Fc receptor present on B cells, is a negative co-receptor for BCR. Dysfunction of FcγRIIb contributed to excessive B cell responses and autoimmunity. Mice on the nonautoimmune C67BL/6 background develop spontaneous lupus when the gene for FcγRIIb is deleted [Citation43]. Further, B cells expression of FcγRIIb is abnormally low in SLE leading to inadequate suppression of autoantigen mediated-BCR activation [Citation44]. Therefore, FcγRIIb represents a novel therapeutic target in SLE. The pharmacological co-engagement of FcγRIIb and BCR complex using recombinant antibodies is an approach to induce B cell suppression.
There are two agents targeting CD19, the XmAb5871 which is a bispecific antibody targeting CD19 and FcγRIIb which functions by inhibiting and preserving B cells, and the CD19 chimeric antigen receptors (CARs) engineered receptors on T cells in targeting CD19 leading to B cells depletion.
A bispecific antibody, XmAb5871, also known as obexelimab, was designed to bind FcγRIIb with its Fc domain and with a humanized Fv region against CD19. The Fc domain was engineered to have >400 –fold increase affinity for FcγRIIb compared to native IgG1 Fc [Citation45]. XmAb5871 suppress B cell activation and also inhibited B cell proliferation induced by other B cells activation signals such as IL-4, BAFF and lipopolysaccharide (LPS). In addition, co-engagement of BCR and FcγRIIb can also inhibit APC function, T cell stimulation, as well as toll-like receptor (TLR) 9 mediated signal [Citation29,Citation46]. XmAb5871 Fc domain targets only FcγRIIb on B cells but it does not bind to the activating receptor, FcγRIII on natural killer (NK) cells, hence B cell depletion though the antibody-dependent cell-mediated cytotoxicity (ADCC), the common anti-CD19 pathway, would not occur in patients treated with XmAb5871 [Citation20]. Therefore, treatment with XmAb5871 might be a good approach in SLE.
To date, there is only one randomized, double-blinded, placebo-controlled phase II trial of XmAb5871 currently ongoing (NCT02725515) aiming to determine the ability of XmAb5871 in maintaining SLE disease activity improvement achieved by a brief course of steroid therapy.
Emerging immunotherapy approach known as CARs-T cell therapy uses engineered receptors bind onto T cell surface which then able to target B cells. The process includes an adoptive cell transfer (ACT) collecting method using T cells from patients and reengineered with CARs. Then CARs-T cells are infused back into the patient recognizing and attacking the cells that have the targeted antigen on their surface [Citation47].
Engineered T cell receptors which target CD19+ B cells, anti-CD19 CARs, are able to deplete CD19+B cells. Further, chimeric autoantibody receptor (CAAR) have been successfully cloned into T cells, leading to selectively killing of autoreactive B cells while sparing healthy B cells [Citation48]. CAAR with extracellular domain of desmoglein (Dsg) 3 was successfully used in pemphigus vulgaris mouse model to engineer T cells to kill autoimmune B cells. Dsg 3 CAAR-T did not lyse the human Dsg3- keratinocytes suggesting that Dsg3 CAAR-T cause no toxicity to non-autoreactive cells. Therefore, CAAR-T cells could be developed as a targeted approach in SLE while avoiding general immunosuppression.
In 2017, two anti-CD-19 CARs-T cell therapies were approved by FDA, one for treatment of acute lymphoblastic leukemia (ALL) in children, and another for adults with advance lymphomas. To date, there is only one on ongoing clinical trial, an open-labeled, uncontrolled, single-arm phase I pilot study in CD19+ SLE patients in China (NCT03030976).
CD19 targeting therapies are still under development in SLE and their effects still yet to be known.
3.1.2. Anti-CD20 targeting therapies
CD20 is an activated-glycosylated phosphoprotein only expressed on B cells from pre-B cells to memory B cells, but not expressed neither by pro-B cells, antibody-producing plasma cells or T cells. Consequently, targeting CD20 spares the B cell precursor, pro-B cells and plasma cells, removing the autoreactive memory B cells and reconstituting the population of naïve transitional B cells [Citation49].
Three agents which target CD20 have been investigated in SLE. Rituximab, a chimeric antibody which depletes B cells via ADCC, complement mediated cytotoxicity (CDC), apoptosis and antibody-dependent phagocytosis (ADP). Ocrelizumab a humanized anti-CD20 monoclonal antibody, and the newly developed glycoengineered anti-CD20 antibody obinituzumab with strong affinity to NK cells.
Rituximab is a CD20 IgG1 type 1 chimeric monoclonal antibody that specifically targets the CD20 molecule on the surface of B cells and destroys B cells. It was first approved by the FDA for the treatment of B-cell lymphomas [Citation50]. The Fab domain of rituximab binds to the CD20 antigen while the Fc domain recruit immune effector cells such as monocytes, macrophages, neutrophils and NK cells, resulting in B cell death. Rituximab-opsonized B cells are subject to attack and killed by four major independent mechanisms: CDC, ADCC, direct ability to induce apoptosis, and ADP [Citation51,Citation52]. In addition, binding of rituximab to B cells may act as decoy immune complexes that efficiently divert monocytes or macrophages with tissue-associated immune complexes, and, therefore, reducing inflammation and tissue damage.
Numerous open-label studies using rituximab in SLE patients reported favorable results. A systemic review of the use of rituximab in 188 patients from 35 articles (9 uncontrolled studies and 26 case reports) revealed that 91% patients had a significant improvement in at least one systemic SLE manifestation. Adverse events, most frequently infections, were reported in 23%, The global analysis of all cases supported the use of rituximab in severe, refractory SLE cases [Citation30]. Moreover, a systematic analysis of reports that document outcomes of rituximab treatment for refractory lupus nephritis suggests that rituximab effectively induces remission of lupus nephritis in patients who have not achieved remission with standard therapies [Citation53].
Further, the European registries have reported good efficacy and safety of rituximab in refractory renal and non-renal SLE manifestations with a high response rate at 6 and 12 months [Citation54–57].
Two randomized, double-blind, placebo-controlled trials evaluating the efficacy and safety of rituximab versus placebo in SLE patients have been completed. The Exploratory Phase II/III SLE Evaluation of Rituximab (EXPLORER, NCT00137969), was performed in moderate to severe active non-renal SLE patients receiving baseline immunosuppressive drugs. Despite of significant improvement in anti-dsDNA antibody titers and complement levels, there was no significant difference in clinical response, measured as BILAG scores, between the two treatment arms [Citation58]. Similar results were obtained from the Lupus Nephritis Assessment with Rituximab (LUNAR, NCT00282347) a phase III, randomized, controlled trial in patients with active proliferative lupus nephritis receiving steroids and MMF [Citation59].
There were no statistically significant differences in the numbers of adverse events and serious adverse events between rituximab group and the control group in both EXPLORER and LUNAR. Several reasons might have contributed to the disappointed outcomes of these two trials including the background treatment with immunosuppressive agents, the high dose of corticosteroids or the relatively short duration of trials.
A phase III, open label randomized controlled trial of rituximab and MMF without oral steroids in patients with lupus nephritis (RITUXILUP, NCT01773616) started in 2015 in United Kingdom (UK) and stopped due to unknown reasons. The pilot study conducted prior RITUXILUP, has proven the ability of the rituximab-based regimen to treat lupus nephritis in the absence of steroids [Citation60].
One main concern in the treatment with rituximab in SLE patients is the production of human anti-chimeric antibody (HACA). HACA titers correlated with poor B cell depletion. In addition, development of HACA could decrease the drug effect and render subsequent courses of rituximab ineffective [Citation61].
Rituximab can trigger a sequence of events that exacerbate the disease in some patients with SLE. The post-rituximab flares, also known as the “BAFFling effects, are characterized by high levels of anti-dsDNA antibodies associated with elevated circulating BAFF levels and a high proportion of plasmablasts in the B cell pool. BAFF perpetuates autoreactive B cells and also stimulates T follicular helper (TFH) cells [Citation62]. CD20 negative cells of the B cell linage such as plasma cells, are not directly affected by rituximab and these cells may contribute to maintenance of disease in some patients.
To date, there is one phase II, randomized, controlled trial, Rituximab Objective Outcome Measures Trial in SLE, (ROOTS, NCT03054259) to evaluate the efficacy of rituximab in SLE patients with skin disease and arthritis.
The humanized anti-CD20 monoclonal antibody, ocrelizumab, has been approved by FDA in 2017 as a treatment for multiple sclerosis. Ocrelizumab has higher ADCC activity and lower CDC effects compared to rituximab [Citation63], therefore, it might be able to modulate pathogenic response more effectively than rituximab. Ocrelizumab does not produce HACA [Citation63] and thus could be a more favorable agent than rituximab.
To date, there is only one completed phase III, randomized, double-blind, placebo-controlled trial of ocrelizumab in SLE that in patients with class III/IV lupus nephritis who were receiving standard care of cyclophosphamide or MMF (BELONG, NCT00626197) [Citation64]. The trial was designed to continue for more than 2 years, but it was stopped prematurely due an imbalance in serious infections between both ocrelizumab-treated patients versus placebo-treated patients in the subgroup of patients receiving background MMF. The analysis of 223 patients receiving ≥32 weeks of treatment indicated that the overall renal response rates with ocrelizumab were numerically but no statistically significant superior to those placebo group.
Another phase III trial with ocrelizumab was initiate in non-renal SLE patients, BEGIN (NCT00539838), but it was terminated early and results were not reported.
Obinutuzumab is a glycoengineered fully humanized anti-CD20 IgG1 type 2 monoclonal antibody binds to an epitope on CD20 that could partially overlap with the epitope recognized by rituximab [Citation65]. Obinutuzumab has greater affinity than rituximab for NK cells leading to a high ADCC activity [Citation66,Citation67]. Recent reports have shown that obinutuzumab is more efficient than rituximab at inducing B cell cytotoxicity in in vitro whole blood assays in SLE and rheumatoid arthritis (RA) patients [Citation68]. A phase II randomized, double-blind, controlled trial comparing the efficacy and safety of obinuzutumab plus MMF/mycophenolic acid (MPA) in proliferative lupus nephritis patients is currently ongoing (NCT02550652).
Finally, some case reports have shown the efficacy of ofatumumab, a fully humanized type 1 IgG1κ monoclonal anti-CD20 antibody in patients with severe SLE or lupus nephritis [Citation69,Citation70]. Ofatumumab binds to CD20 with higher affinity and activates CDC and ADCC more effectively than rituximab in in vitro assays and causes prolonged B cell depletion in animal studies [Citation71]. Clinical studies reported the efficacy of this drug in the treatment of RA and relapsing-remitting multiple sclerosis [Citation72–74], some case reports have shown its efficacy in severe SLE or lupus nephritis patients [Citation69,Citation70]. To date there is no clinical trial evaluating the efficacy of ofatumumab in SLE.
In summary, the strong depleting effects of rituximab and the ability of producing HACA might be the major causes of its failure in treating SLE. Although, ocrelizumab is supposed has lesser extent in B cells depletion, the severe adverse events related to its stronger ADCC effects could not be neglected. Obinutuzumab with strong affinity to NK cells still remains under investigation.
3.1.3. Anti-CD22 targeting therapies
CD22, also known as sialic acid-binding Ig-like lectin 2 (Siglec-2) or B-lymphocyte cell adhesion molecule (BL-CAM) [Citation75] is a B-cell-restricted membrane co-receptor expressed on the surface of pro-B cell. It is present throughout the most of B cell development, highly expressed on naïve B cells, but not expressed on plasmablasts and plasma cells [Citation76].
The first CD22-specific treatment tested in clinical trials of SLE is epratuzumab, a monoclonal antibody which inhibits B cell proliferation, enhances the normal inhibitory function of CD22 on the BCR and limits the extent of BCR signaling reducing B cell stimulation [Citation77].
Two phase III, randomized, double-blind, placebo-controlled multicenter studies, the ALLEVIATE 1 (NCT00111306) and ALLEVIATE 2 (NCT00383214) were carried out in patients with moderately to severely active SLE but both trials were discontinued prematurely because of interruption in drug supply. The exploratory pooled analysis of the data showed total BILAG scores lower in both epratuzumab arms (360 mg/m2 and 720 mg mg/m2) versus placebo and similar incidence of adverse events. These efficacy and safety profile of epratuzumab for SLE treatment supported its continued development for SLE [Citation31]. These results are consistent with the first clinical trial of epratuzumab in SLE, a phase II, open-label, non-randomized, single-center study including 14 patients with moderately active SLE. Epratuzumab was safe and well-tolerated with few significant adverse events [Citation78].
Promising results were published from the phase IIb randomized, double-blind, placebo-controlled study EMBLEM (NCT00624351) [Citation79]. The study evaluated several doses of epratuzumab in 227 moderately to severely active SLE patients using a composite end-point, the BICLA. At 12 weeks, the responder rate in the placebo arm (21.1%) was lower than in the epratuzumab arm (45.9%), but the overall test of treatment effect was not statistically significant. The exploratory pairwise analysis of responder rates demonstrated statistically significant clinical improvements in patients receiving a cumulative dose of 2400 mg epratuzumab versus placebo. No decreases in Ig levels outside normal ranges were observed during EMBLEM. Unlike rituximab, epratuzumab only partially reduces B-cell numbers and, therefore, it is possible that this agent may not be associated with decreases in Ig levels or an increased risk of infection.
Extension study of long-term safety and efficacy of epratuzumab has been done in patients who enrolled EMBLEM up to 3.2 years. Epratuzumab was well-tolerated with sustained improvements in disease activity and health-related quality of life (HRQOL), as well as reduction of steroid doses [Citation80]. Two phase III randomized, double-blind, placebo-controlled studies, the EMBODY 1 (NCT01262365) and EMBODY 2 (NCT01261793) assessed the efficacy and security of two 2400 mg cumulative dose regimens of epratuzumab versus placebo in moderate to severe active SLE patients. In both of these trials, the observed immunological effects of epratuzumab treatment were as expected leading to reductions in the levels of Ig and CD22 with the number of B cells in peripheral blood decreasing by 30–40%. However, treatment with epratuzumab in combination with standard therapy did not achieve statistically significant difference in the primary end point, response rate at week 48 according to the BILAG-BICLA definition, compared with placebo in conjunction with standard therapy [Citation81]. There are several factors that could have influenced the results of EMBODY1/2. First, there was a high discontinuation rate, approximately one-third of patients discontinued the study prior to week 48 and were classified as non-responders. Another important factor is the corticosteroid dosage, about 40% of patients did not reduce their corticosteroids dosage in these trials and another 40% of patients increased their corticosteroid dosage or had missing data. These issues lead to high placebo response rates at the end points. Despite the disappointment in the trials outcome, another phase 3 extension study (EMBODY4) in SLE has been completed and undergone analysis. A summary of clinical trials of therapies targeting B cell surface antigens is shown in .
Table 2. Promising therapies targeting B cell surface antigens for SLE currently under investigation.
In summary, epratuzumab has been undergone several clinical trials and the long-term study suggests its potential in SLE treatment.
3.2. Targeting B cell survival factors
The BAFF, also known as B lymphocyte stimulator, and APRIL, are two related members of the TNF ligand superfamily of proteins that promote B cell survival, differentiation and maturation, play a key role in the development of autoimmunity, especially in SLE [Citation82].
BAFF is mainly expresses by monocytes, macrophages and activated T cells and can remain in a membrane bound form or released as a soluble form after cleavage by furin [Citation83]. Only the processed soluble BAFF form is required for B cell development [Citation84]. In contrast, APRIL normally exists in a soluble form only [Citation85].
BAFF binds to three receptors, the BAFF receptor (BAFF-R), mainly expressed in immature B cells, the transmembrane activator and cyclophilin ligand interactor (TACI), expressed in memory B cells, or the B cell maturation antigen (BCMA) expressed in plasmablasts and plasma cells. APRIL forms heterotrimers with BAFF and enhances BAFF-mediated cell activation [Citation86]. APRIL binds to TACI, BMCA and proteoglicans.
Development and survival of immature B cells and naïve B cells mostly depend on BAFF. On the other hand, APRIL tends to interact more avidly with antigen-experienced B cells than BAFF, binding to BCMA and promoting long-lived plasma cell survival [Citation82,Citation87]. BAFF can also promote T cell activation, proliferation and differentiation. The balance between BCMA and BAFF-R signaling may control the development of TFH cells indicating that the BAFF/APRIL can also regulate autoimmunity via expansion of TFH cells [Citation88].
Animals studies revealed that knockout of BAFF in lupus-prone mice causes a reduction in disease severity and mortality, and BAFF transgenic mice developed severe lupus disease [Citation89]. In SLE patients, serum concentrations of BAFF and APRIL were higher compared to those in healthy individuals [Citation90], and positively correlation was found between plasma BAFF levels and SLE disease activity. All these findings support BAFF/APRIL system as a candidate for therapeutic targeting in SLE [Citation91].
3.2.1. BAFF/APRIL system blockers
Selective blockers of BAFF and APRIL have been investigated in clinical trials for SLE. These agents inhibit autoreactive B cell activation and autoantibody productions consequently should improve autoimmunity
Belimumab is a fully humanized monoclonal antibody that specifically binds to soluble trimeric BAFF, preventing his interaction with receptors, thus inhibiting B cell maturation and survival. Recent report suggests its binding to membrane-bound BAFF too [Citation92].Two large double-blind phase II, multicenter, randomized placebo-controlled trials, BLISS-52 and BLISS-76 evaluated the efficacy of belimumab in patients with SLE receiving standard therapies (NCT00424476, NCT00410384). Both studies have reported that belimumab plus standard therapy has significantly improved SRI response rate, reduced SLE disease activity and severe flares with similar rates of adverse events compared to placebo group [Citation32,Citation93]. A pooled analysis of these two trials indicated that belimumab has greater therapeutic benefit than standard therapy alone in patients with higher disease activity, anti-dsDNA positivity, low complement or corticosteroid treatment at baseline [Citation94]. Post hoc analysis indicates that belimumab treatment improved overall SLE disease activity in the most common musculoskeletal and mucocutaneous organ domains [Citation95]. In patients with renal involvement, pooled subgroup analysis showed greater improvement in patients receiving belimumab that in those on placebo groups [Citation96]. Belimumab was well-tolerated and there was no significant difference in the frequencies of serious adverse events between belimumab and placebo groups [Citation97].
Further, a phase III randomized placebo-controlled study in patients with SLE conducted in China, Japan and South Korea showed significantly improved disease activity and reduction in prednisone use, with no new safety issues (NCT01345253) [Citation98]. The phase III study (EMBRACE), which evaluates the efficacy and safety of belimumab in black race, failed to meet the primary endpoint, SRI response rate with modified SLEDAI-2000 (SLEDAI-2K) scoring for proteinuria at week 52, suggests that belimumab has different effects on different races. However, the significant improvement observed in subgroups with high disease activity (HDA) supports belimumab treatment in black SLE patients with high HDA [Citation99].
These results highlighted the efficacy and tolerability of belimumab as a novel therapeutic agent for SLE. The FDA approved belimumab (Benlysta) in 2011 for SLE, then it was approved by the National Institute for Health and Care Excellence (NICE) in 2016 and by the Japanese Ministry of Health, Labour and Welfare in September 2017.
However, the efficacy of belimumab has not been evaluated in patients with major organ involvements including severe active lupus nephritis or severe active central nervous system (CNS) lupus and it has also not yet been studied in combination with other biologics or intravenous cyclophosphamide. To date there are numerous ongoing clinical trials of belimumab in SLE patients ().
Table 3. Current ongoing trials of belimumab in SLE.
As previously mentioned, BAFFling effects occur after rituximab treatment. These effects are characterized by high levels of anti-dsDNA antibodies associated with elevated circulating BAFF levels and a high proportion of plasmablasts in the B cell pool [Citation100]. BAFFling effects might be diminished or eliminated by using belimumab after rituximab treatment. To date, there are 2 ongoing studies evaluating the efficacy of co-administration of rituximab and belimumab: a phase II interventional trial, synergetic B cell immodulation in SLE (SYNBloSe, NCT02284984); and a phase III randomized, double-blind, placebo-controlled trial of belimumab administered in combination with rituximab in SLE patients (BLISS-BELIEVE, NCT03312907). A phase II study about rituximab plus cyclophosphamide followed by belimumab in lupus nephritis patients has been completed and undergone analysis (CALIBRATE, NCT02260934).
Other anti-BAFF agents are also being assessed in SLE patients and are currently undergoing clinical testing.
Tabalumab, a human IgG 4κ anti-BAFF monoclonal antibody that targets both membrane-bound and soluble bound BAFF [Citation101]. The membrane-bound BAFF is at least 50-fold more active than the soluble BAFF, thus a potential greater clinical response might be achieved by inhibiting membrane-bound BAFF, or both forms of BAFF. Remarkably, tabalumab underwent neither phase I nor phase II evaluation in SLE but plunged directly into two separate phase III studies, the ILLUMINATE-1 (NCT01196091) and ILLUMINATE-2 (NCT01205438) [Citation102,Citation103]. ILLUMINATE-1 did not meet the SRI-5 primary endpoint at week 52 [Citation102]. ILLUMINATE-2 achieved SRI-5 primary endpoint but failed to meet the multiplicity-controlled secondary endpoints including the time-to-first severe SLE flare on the SELENA-SLEDAI Flare Index, corticoid sparing effects and fatigue at week 52 [Citation103]. The pharmaceutical company discontinued the development of this drug for SLE.
Blisibimod, also known as A-623 or AMG 623, is a human peptibody which binds to both soluble and cell membrane-bound forms of BAFF [Citation104]. A phase II, randomized, placebo-controlled trial of blisibimod in SLE, PEARL-SC (NCT01162681) confirmed its favorable safety and tolerability compared with placebo but failed to meet the primary endpoint SRI-5 at week 24 [Citation105]. In this study, significant increases from baseline in serum concentrations of complement C3 and C4, and significant reductions in concentration of anti-dsDNA autoantibodies observed with blisibimod compared with placebo support the continuing development of blisibimod as a therapy for patients with SLE.
Recently, a phase III randomized, double-blind, placebo-controlled study, CHABLIS-SC1 (NCT01395745) of blisibimod in patients with active SLE failed to meet its primary end-point of SRI-6. However, blisibimod had successfully reduced the usage of steroid, levels of proteinuria and biomarker responses suggesting that blisibimod might have important clinical benefits [Citation106].
Another phase III study of blisibimod in SLE patients with or without nephritis, the CHABLIS7.5 is still ongoing (NCT02514967). A long-term safety PEARL-SC (NCT01305746) extension trial evaluating the effects of blisibimod on renal and inflammation biomarkers in SLE patients has been completed and reported decreased in proteinuria, anti-ds-DNA antibodies, number of peripheral CD19+/CD20+ B cells, serum levels of IgG and IgM and increased glomerular filtration rate (GFR) compared to placebo group, with similar safety profile [Citation107].
Atacicept, previously referred to as TACI-Ig, is a fully human recombinant fusion protein containing the extracellular domain of TACI receptor joined to a human IgG1 the Fc domain. Atacicept binds to both BAFF and APRIL and inhibits activation of TACI-mediated signaling. A multicenter, Phase Ia double-blind placebo-controlled trial of atacicept versus placebo in patients with mild to moderate SLE demonstrated that treatment with atacicept resulted in dose-dependent reductions in Ig levels and sustained dose-related reduction of total B cells compared to placebo. There were no differences in the frequency or type of adverse events in patients treated with atacicept [Citation108]. However, a II/III randomized, double-blind, placebo-controlled study of atacicept in lupus nephritis patients in combination with MMF and corticosteroids (NCT00573157) was prematurely terminated after the enrollment of six patients due to an unexpected decline in serum IgG and the occurrence of serious infections [Citation109]. Moreover, a phase II/III randomized, double-blind, placebo-controlled trial APRIL-SLE (NCT00624338) was designed to assess whether atacicept could prevent flares in patients with moderate-to-severe SLE administered together with a course of corticosteroids. Patients were randomized to 75 mg or 150 mg of atacicept or placebo group. Two fatal infections in the 150 mg atacicept group led to premature termination of this group. There was no significant difference in 75 mg atacicept group compared to the placebo group in primary endpoint, the proportion of patients experiencing at least one flare of BILAG A or B, as well as in secondary endpoint or time to first flare [Citation33].
A 24-week, randomized, double-blind, placebo-controlled phase IIb study compared the efficacy and safety of two doses of atacicept (75 and 150 mg) with placebo in patients with active SLE receiving standard treatment, ADDRESS II (NCT01972568). In predefined subpopulations with HDA at baseline and/or serologically active disease statistically significant improvements in the SRI-4 and SRI-6 response rates were seen with atacicept versus placebo. The risks of serious adverse events and serious or severe infection were not increased with atacicept as compared with placebo [Citation110]. A long-term extension of ADDRESS II was evaluated the long-term safety and tolerability of atacicept in SLE patients (NCT02070978) and has been successfully extended for another 24 weeks. The results of this trial indicate that rates of flares continued to be low in atacicept-treated patients [Citation111].
A novel soluble, fully human recombinant fusion protein, RCT-18, binds and neutralized the activity of BAFF and APRIL. This protein is constructed with the soluble extracellular BAFF/APRIL domain of TACI receptor and the Fc portion of human IgG. Compared to atacicept, RCT-18 reserves the N-terminal fragment of extracellular TACI and increases the binding capacity of TACI to BAFF [Citation34]. Currently, a phase IIb randomized, double-blind, placebo-controlled trial to determine the doses of RCT-18 in SLE patients is in progress (NCT02885610).
Overall, belimumab, tabalumab and blisibimod target BAFF. However, tabalumab and blisibimod failed to achieve endpoints and worsen the patients’ condition. Atacicept and RCT-18 target both BAFF and APRIL, while atacicept has been recognized to reach the end point in trials, it is not recommended due to the fatal adverse effects. RCT-18 is still under investigation.
summarized the clinical trials targeting B cell survival factors in SLE patients.
Table 4. Therapies targeting B cell survival factors, cytokines and co-stimulatory molecule for SLE currently under investigation.
3.3. Targeting cytokines
Cytokines are secreted by different immune cells and modulate the activation and/or functions of several target cells in the immune systems. Interferons and some IL have an active role in the pathogenesis of SLE and can contribute significantly to the immune imbalance in the disease. Cytokine-based therapies could indirectly target B cells in SLE patients.
3.3.1. IL-6 and its receptor
IL-6 is a pleiotropic cytokine produced mainly by macrophages and monocytes, but also by T and B cells. IL-6 plays an important role in immune regulation and inflammation [Citation112].
The receptor for IL-6 consist of two functional proteins, a 80 kDa glycoprotein (gp) namely IL-6R and gp130. IL-6R is the ligand specific binding component while gp130 is a shared receptor component responsible for transmitting signals of IL-6 related cytokines.
Data from animal studies indicated an association between IL-6 and SLE progression and results for human studies suggest that IL-6 plays a critical role in the B cell hyperactivity an immunopathology of SLE [Citation112]. In SLE patients, serum concentration of IL-6 was substantially elevated compared to healthy subjects [Citation113]. Treatment with hydroxychloroquine dramatically decreases IL-6 levels in SLE patients [Citation114]. Moreover, neutralizing antibodies against IL-6 inhibits autoimmune response including spontaneous polyclonal antibody production by SLE monocytes and lymphocytes in vitro [Citation115]. Taken together, targeting IL-6 signaling may improve SLE by indirectly downregulate the B cell differentiation into plasma cells and decrease IL-6 levels.
Sirukumab, formerly called CNT0136, is a human anti-IL-6 monoclonal antibody that binds to IL-6 with high affinity ad inhibits IL-6-mediated signal transducer and activator of transcription 3 (STAT-3) phosphorylation attenuating the biological activity of IL-6. Results from a phase I randomized, double-blind, placebo-controlled study of sirukumab in patients with CLE or SLE (NCT01702740) reported that sirukumab infusions were generally well tolerated in patients with mild, stable, active disease [Citation35]. Another phase II randomized, double-blind, placebo-controlled study evaluated efficacy and safety of sirukumab in patients with class III or class IV lupus nephritis (NCT01273389) showed that the addition of sirukumab to the treatment regimen did not result in an overall improvement in proteinuria at week 24 and 47.6% of patients in sirukumab group had more than 1 serious adverse events through week 40 [Citation116]. There is no ongoing trial on sirukumab in SLE.
Tociluzimab is humanized anti- IL-6R that prevents the binding of IL-6 to both soluble and membrane-bound IL-6R. Only one phase I, open-labeled study of tocilizumab in SLE patients has been done (NCT00046774). The preliminary data showed significant improvements of disease activity after treatment with tocilizumab, but the increased risk of infection and neutropenia observed may limit its use in SLE [Citation36].
Vobarilizumab, also known as ALX-0061 is an anti-IL-6R antibody with longer half life than tocilizumab composed of two heavy chains. One chain targets human serum albumin and is designed to prolong the molecules half-life by up to 14 days; the other chain targets the soluble IL-6R. Ablynx, the developer of vobarilizumab, failed to meet the primary endpoints in the phase II randomized, double-blind, placebo-controlled trial in moderate to severe SLE patients (NCT02437890) and serious adverse events were reported in 2 percent in vobarilizumab group [Citation117] suggesting that it has limiter potential in treating SLE.
In summary, sirukumab targets IL-6 while tocilizumab and vobarilizumab target IL-6R. The overall effect of these biologics is the inhibition of terminal differentiation of B cell into plasma cells. Sirukumab did no improvement in proteinuria in lupus nephritis patients whereas tocilizumab leads to a significant improvement in disease activity in SLE.
3.3.2. Type 1 interferons
Type 1 interferons (IFN-I), and particularly IFN-α, are central mediators in the pathogenesis of SLE [Citation118]. Cell signaling by all IFN-I, including IFN-α, IFN-β, IFN-ε, IFN-κ, and IFN-ω, is mediated by IFN-I-α/β/ω receptor (IFNAR).
IFN-α is produced by B cells and plasmacytoid dendritic cells (pDCs) and participated in various pathogenic pathways in SLE [Citation119]. IFN-α induces BAFF, which then indirectly support B cell differentiation and Ig class switching to generate potentially pathogenic autoantibodies [Citation120]. Natural IFN-α-producing cells, pDC, have been found in the skin biopsies of CLE patients, with local productions of IFN-α [Citation121]. The presence of IFN regulated genes correlated with anti-dsDNA antibodies and SLE clinical manifestations [Citation122]. Moreover, development of SLE has been reported in patients receiving IFN-α therapy for a variety of conditions [Citation123]. Therefore, blockade of IFN-α not only regulate B cells, but also able to diminish its supports in generating autoantibodies.
Rontalizumab is a humanized IgG1 anti-IFN-α monoclonal antibody which prevents signaling through IFN-IR by neutralizing all known subtypes of human IFN-α but does not bind to IFN-β or IFN-ω. A phase I randomized, double-blind, placebo-controlled trial has confirmed its preliminary safety, pharmacokinetics profile and pharmacodynamics effects in patients with mild active SLE (NCT00541749) [Citation37]. A multicenter, randomized, placebo-controlled phase II efficacy and safety trial on rontalizumab (ROSE; NCT00962832) did not met the primary (BILAG-2004) or secondary (SRI) end points at week 24. However, a post hoc analysis suggested potential benefit in a small subset of patients with a low baseline IFN regulated genes [Citation124].
Sifalimumab, formerly MEDI-545, is a fully human IgG1κ monoclonal antibody that binds to IFN-α with high affinity and prevents IFN-α signaling through the type I IFN-R. Sifalimumab inhibits most but not all type I IFNα subtypes, but does not inhibit β or δ IFNs. Clinical trials in SLE patients have established the safety profile of sifalimumab have suggested favorable effects on clinical outcome measures [Citation125]. IFN target neutralization was significantly less in SLE patients with moderate to severe disease than in those with mild disease [Citation125,Citation126], probably reflecting an increased contributions of types Iβ and δ IFNs to the target signature in patients with moderate to severe SLE. A phase IIb, randomized, double-blind, placebo-controlled study demonstrated greater efficacy with sifalimumab than placebo in patients with moderate to severe active SLE and an inadequate response to standard-of-care treatments (NCT01283139) [Citation127]. The broad-based improvement measured in both SLE composite end points (SRI-4, BICLA, BILAG-based Composite Lupus Assessment) and individual organ systems (CLASI, joint counts) supports evidence that IFN-α plays an important role in SLE pathogenesis. Another three completed phase II studies on sifalimumab in SLE patients (NCT00979654, NCT00657189, NCT01031836) reported to meet primary and some secondary end points but the treatment effects were modest, and the developer of sifalimumab had made the decision to discontinue its development in favor of another IFN-I inhibitor, anifrolumab.
Anifrolumab, also known as MEDI-546, is a fully human, IgG1κ monoclonal antibody that binds to the IFNAR, resulting in IFN-stimulated gene transcription [Citation128]. Both rontalizumab and sifalimumab have specificity only for IFNα, leaving other type I IFNs unaffected and able to bind IFNAR. On the other hand, anifrolumab, has the ability to block the entire IFN-I signaling through blocking type I IFN autoamplification loop and proinflammatory cytokine induction [Citation129]. Therefore, it is considered as a promising therapeutic biologic for SLE as one of its major pathogenic mechanisms is chronic dysfunctional type I IFN signaling [Citation118]. A phase II randomized, double-blind, placebo-controlled study of anifrolumab, in SLE patients with moderate to severe SLE (NCT01438489) met the primary endpoint SRI-4 at week 24 with a sustained reduction of oral corticosteroids [Citation38]. To date, three phase III studies on anifrolumab in active SLE patients are ongoing (NCT02446899, NCT02446912, NCT02794285).
In summary, rontalizumab which targets IFN-α, sifalimumab and anifrolumab which target type I IFN-R have a good response and little adverse events in clinical trials. Blocking IFNAR is a promising and novel therapeutic approach for patients with SLE whose disease does not respond to currently available therapies.
Current clinical trials targeting cytokines are shown in .
3.4. New treatments targeting co-stimulator molecules
3.4.1. Cd40l
Co-stimulation pathways involves mutual exchange of information and signaling and play an essential role in initiating, perpetuating and attenuating the proinflammatory immune response. The costimulatory molecule CD40 and its ligand CD40L, the CD40-CD40L system has pleiotropic effects in a variety of cells and biological processes.
CD40L has been shown to be an important immune-inflammatory modulator such it is a credible candidate for pharmacological intervention. CD40L is essential for the growth, differentiation, resistance to apoptosis, and effector functions of B cells [Citation130]. CD40L is widely expressed on naïve and activated CD4+ T cells and platelets [Citation131]. CD40–CD40L engagement is essential for normal T cell/B cell functional interactions of CD8+ T cell responses, Ig class-switching and induction of dendritic cell maturation [Citation132–134]. Thus, targeting CD40L on T cells would eventually lead to diminish the function of B cells.
CD40L was reported to be overexpressed on T cells of female lupus patients [Citation135,Citation136] and CD40-CD40L system has an important role in the production of pathogenic autoantibodies and tissue injury in lupus nephritis [Citation137]. These characteristics favor CD40L as a therapeutic target in SLE patients.
Two monoclonal antibodies against CD40L have been evaluated in SLE, ruplizumab and toralizumab. Ruplizumab, BG9588, is a humanized monoclonal anti-CD40L IgG1 antibody which blocks antigen-specific IgG responses in nonhuman primates (baboons and rhesus monkeys) with a variety of T-dependent antigens. Ruplizumab was analyzed in a phase II study in patients with proliferative lupus glomerulonephritis (NCT0001789) showing a reduction in proteinuria, hematuria, and anti-dsDNA titers and an increase serum C3 levels. The study was prematurely terminated after 2 patients experienced myocardial infarction [Citation137]. Toralizumab, another anti-CD40L IgG1 antibody, was also associated with thromboembolic events [Citation138].
A new monoclonal antibody against CD40L, dapirolizumab pegol CPD7657, has been developed. It is an anti-CD40L Fab’ antibody fragment conjugated to polyethylene glycol (PEG). This molecule does not have Fc fragment and lack the ability to bind the platelet receptor FcγIIa, which limits the risk of increased platelet aggregation. In two phase I trials in SLE patients, no thromboembolic events were recorded and good safety profile reported [Citation39,Citation139]. There is one ongoing phase II study evaluating efficacy and safety of dapirolizumab pegol in moderate to severe SLE patients (NCT02804763) ().
In summary, the development of ruplizumab and toralizumab has been discontinued due to the severe adverse events. Dapirolizumab has lesser extent in platelet aggregation and hence no thromboembolic event has been reported. Its potential in treating SLE is still under investigation.
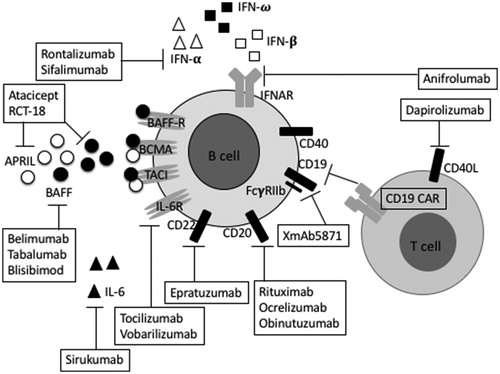
illustrates the current approaches to B cell-directed therapies in SLE.
Figure 1. Approaches to target B cells in SLE. One of the current strategies for SLE treatment include therapeutic modulation of B cell surface antigens (CD19, CD20 and CD22), B cell survival factors (BAFF, APRIL), cytokines (IL-6 and IFN) and their receptors (IL-6R and IFNAR) and co-stimulatory molecule (CD40L). Agents directly targeting on B cell surface antigens include: XmAb5871 which blocks CD19 and FcγRIIb on B cells and hence inhibits B cell functions, on the other hand engineered T cell receptors on T cell, CD19 CARs, blocks CD19 on B cell. Rituximab, ocrelizumab and obinutuzumab block CD20 and epratuzumab blocks CD22. The most developed approach which indirectly target B cells is inhibition of B cell survival factors. Belimumab, tabalumab and blisibimod block BAFF (closed circles). Atacicept and RCT-18 block BAFF and APRIL (open circles). Targeting cytokines or co-stimulatory molecule is another strategy to indirectly modulate B cells. Tocilizumab and vobarilizumab block IL-6R while sirukumab blocks directly IL-6 (closed triangles). Anifrolumab blocks IFNAR while rontalizumab and sifalimumab has specificity for blocking IFN-α (open triangles) but not IFN-ω (closed squares) and IFN-β (open squares) which also bind to IFNAR. Finally, dapirolizumab blocks CD40L disrupting CD40–CD40L interactions. APRIL: A proliferation-inducing ligand; BAFF: B-cell activating factor; BAFF-R: B-cell activating factor receptor; BCMA: B-cell maturation antigen; CARs: Chimeric antigen receptors; IL-6: Interleukin-6; IL-6R: interleukin 6 receptor; IFN: Interferon; IFNAR: the type 1 IFN-α/β/ω receptor; TACI: transmembrane activator and CAML interactor
3.5. New trends in targeted therapies
We have described targeting therapies against B cells surface and B cells related cytokines which are located outside of B cells. Recently, researchers started to focus on SLE treatment which targets molecules within the cells. Bruton’s tyrosine kinase (BTK), an important molecule in B cells, has been reported to have multiple pathogenic roles in SLE including B cells differentiation and autoantibodies production [Citation140]. One of the BTK inhibitors, evotrubinib, has been reported to be a promising agent in treating chronic autoimmune disease in mice [Citation141]. Further, BTK inhibition was proven to be effective in improving renal, cutaneous and brain manifestations in a murine model of SLE [Citation142]. BTK inhibitors are now undergoing investigation in phase 1 studies in SLE patients (NCT02537028, NCT03878303). Nevertheless, SLE treatment has jumped into a new era in targeting molecules within the pathogenic cells.
4. Conclusion
SLE is a complex and clinically heterogeneous autoimmune disease involving many immunological pathways. SLE management, particularly refractory disease, represents a major challenge for rheumatologists, and the development of specific targeted therapies became a clinical necessity. Autoreactive B cells are one of the key molecules implicated in the pathogenies of SLE and wide range of biologics targeting B cells have been investigated. However, to date, only belimumab, a fully humanized monoclonal antibody that specifically binds to soluble trimeric BAFF, has been approved as a treatment of adult SLE patients.
One of the most promising biologics to treat SLE treatment is XmAb5871, an antibody engineered designed to bind FcγRIIb with its Fc domain and with a humanized Fv region against CD19. This bispecific antibody inhibits B cell function and proliferation without depleting B cells has also been potential to suppress APC function, T cell stimulation and TLR 9 mediated signaling. Although there CAARs therapies are still under development, in SLE, the strategy of reengineered CAAR-T cells targeting autoantibodies-generating B cells has a great potential to treat SLE while avoiding general immunosuppression. The successful trials in SLE with anifrolumab suggest its benefits in targeting type 1 IFN signaling, one of the most important pathogenetic pathway in SLE, and hence anifrolumab is also considered as a novel therapeutic approach in SLE patients who do not respond to current available therapy.
Although numerous open-label studies have been reported the success of rituximab in treating SLE, the failure of several trials in rituximab and other anti-CD20 biologics suggest that targeting CD20+ B cells may not be an option in SLE patients despite their achievement in leukemia diseases.
Finally, due to the heterogeneity of SLE population the development of personalized targeted therapies will be necessary.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Bernatsky S, Boivin J-F, Joseph L, et al. Mortality in systemic lupus erythematosus. Arthritis Rheum. 2006;54(8):2550–2557.
- Mok CC, Kwok CL, Ho LY, et al. Life expectancy, standardized mortality ratios, and causes of death in six rheumatic diseases in Hong Kong, China. Arthritis Rheum. 2011;63(5):1182–1189.
- Gatto M, Kiss E, Naparstek Y, et al. In-/off-label use of biologic therapy in systemic lupus erythematosus. BMC Med. 2014;12(1):30.
- Thong B, Olsen NJ. Systemic lupus erythematosus diagnosis and management. Rheumatology (Oxford). 2017;56(suppl_1):i3–i13.
- Doria A, Iaccarino L, Ghirardello A, et al. Long-term prognosis and causes of death in systemic lupus erythematosus. Am J Med. 2006;119(8):700–706.
- Munoz LE, Lauber K, Schiller M, et al. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol. 2010;6(5):280–289.
- Nashi E, Wang Y, Diamond B. The role of B cells in lupus pathogenesis. Int J Biochem Cell Biol. 2010;42(4):543–550.
- Mak A, Kow NY. The pathology of T cells in systemic lupus erythematosus. J Immunol Res. 2014;2014:1.
- Kornbluth A. Infliximab approved for use in Crohn's disease: a report on the FDA GI advisory committee conference. Inflamm Bowel Dis. 1998;4(4):328–329.
- Feldmann M. Development of anti-TNF therapy for rheumatoid arthritis. Nat Rev Immunol. 2002;2(5):364–371.
- Sanz I, Lee FE. B cells as therapeutic targets in SLE. Nat Rev Rheumatol. 2010;6(6):326–337.
- Mitka M. Treatment for lupus, first in 50 years, offers modest benefits, hope to patients. JAMA. 2011;305(17):1754–1755.
- Stohl W. Inhibition of B cell activating factor (BAFF) in the management of systemic lupus erythematosus (SLE). Expert Rev Clin Immunol. 2017;13(6):623–633.
- Mikdashi J, Nived O. Measuring disease activity in adults with systemic lupus erythematosus: the challenges of administrative burden and responsiveness to patient concerns in clinical research. Arthritis Res Ther. 2015;17(1):183.
- Nutt SL, Heavey B, Rolink AG, et al. Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature. 1999;401(6753):556–562.
- LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. 2008;112(5):1570–1580.
- Jacob J, Kelsoe G, Rajewsky K, et al. Intraclonal generation of antibody mutants in germinal centres. Nature. 1991;354(6352):389–392.
- Radbruch A, Muehlinghaus G, Luger EO, et al. Competence and competition: the challenge of becoming a long-lived plasma cell. Nat Rev Immunol. 2006;6(10):741–750.
- Cortes-Hernandez J, Ordi-Ros J, Labrador M, et al. Antihistone and anti-double-stranded deoxyribonucleic acid antibodies are associated with renal disease in systemic lupus erythematosus. Am J Med. 2004;116(3):165–173.
- O'Neill SK, Shlomchik MJ, Glant TT, et al. Antigen-specific B cells are required as APCs and autoantibody-producing cells for induction of severe autoimmune arthritis. J Immunol. 2005;174(6):3781–3788.
- Chan OT, Hannum LG, Haberman AM, et al. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J Exp Med. 1999;189(10):1639–1648.
- Santulli-Marotto S, Retter MW, Gee R, et al. Autoreactive B cell regulation: peripheral induction of developmental arrest by lupus-associated autoantigens. Immunity. 1998;8(2):209–219.
- Pistoia V. Production of cytokines by human B cells in health and disease. Immunol Today. 1997;18(7):343–350.
- Duddy ME, Alter A, Bar-Or A. Bar-Or A. Distinct profiles of human B cell effector cytokines: a role in immune regulation? J Immunol. 2004;172(6):3422–3427.
- Koenig KF, Groeschl I, Pesickova SS, et al. Serum cytokine profile in patients with active lupus nephritis. Cytokine. 2012;60(2):410–416.
- Lee J, Kuchen S, Fischer R, et al. Identification and characterization of a human CD5(+) pre-naive B cell population. J Immunol. 2009;182(7):4116–4126.
- Yurasov S, Tiller T, Tsuiji M, et al. Persistent expression of autoantibodies in SLE patients in remission. J Exp Med. 2006;203(10):2255–2261.
- Yurasov S, Wardemann H, Hammersen J, et al. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med. 2005;201(5):703–711.
- Horton HM, Chu SY, Ortiz EC, et al. Antibody-mediated coengagement of FcγRIIb and B cell receptor complex suppresses humoral immunity in systemic lupus erythematosus. J Immunol. 2011;186(7):4223–4233.
- Ramos-Casals M, Soto M, Cuadrado M, et al. Rituximab in systemic lupus erythematosus. A systematic review of off-label use in 188 cases. Lupus. 2009;18(9):767–776.
- Wallace DJ, Gordon C, Strand V, et al. Efficacy and safety of epratuzumab in patients with moderate/severe flaring systemic lupus erythematosus: results from two randomized, double-blind, placebo-controlled, multicentre studies (ALLEVIATE) and follow-up. Rheumatology (Oxford). 2013;52(7):1313–1322.
- Navarra SV, Guzman RM, Gallacher AE, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377(9767):721–731.
- Isenberg D, Gordon C, Licu D, et al. Efficacy and safety of atacicept for prevention of flares in patients with moderate-to-severe systemic lupus erythematosus (SLE): 52-week data (APRIL-SLE randomised trial). Ann Rheum Dis. 2015;74(11):2006–2015.
- Chen X, Zhao Q, Hou Y, et al. Pharmacokinetics, pharmacodynamics, short term efficacy and safety of RCT-18, a novel BLyS/APRIL fusion protein, in patients with rheumatoid arthritis. Br J Clin Pharmacol. 2016;82(1):41–52.
- Szepietowski JC, Nilganuwong S, Wozniacka A, et al. Phase I, randomized, double-blind, placebo-controlled, multiple intravenous, dose-ascending study of sirukumab in cutaneous or systemic lupus erythematosus. Arthritis Rheum. 2013;65(10):2661–2671.
- Illei GG, Shirota Y, Yarboro CH, et al. Tocilizumab in systemic lupus erythematosus: data on safety, preliminary efficacy, and impact on circulating plasma cells from an open-label phase I dosage-escalation study. Arthritis Rheum. 2010;62(2):542–552.
- McBride JM, Jiang J, Abbas AR, et al. Safety and pharmacodynamics of rontalizumab in patients with systemic lupus erythematosus: results of a phase I, placebo-controlled, double-blind, dose-escalation study. Arthritis Rheum. 2012;64(11):3666–3676.
- Furie R, Khamashta M, Merrill JT, et al. Anifrolumab, an anti–interferon-α receptor monoclonal antibody, in moderate-to-severe systemic lupus erythematosus. Arthritis Rheumatol. 2017;69(2):376–386.
- Chamberlain C, Colman PJ, Ranger AM, et al. Repeated administration of dapirolizumab pegol in a randomised phase I study is well tolerated and accompanied by improvements in several composite measures of systemic lupus erythematosus disease activity and changes in whole blood transcriptomic profiles. Ann Rheum Dis. 2017;76(11):1837–1844.
- Mei HE, Schmidt S, Dörner T. Rationale of anti-CD19 immunotherapy: an option to target autoreactive plasma cells in autoimmunity. Arthritis Res Ther. 2012;14(Suppl 5):S1.
- Fearon DT, Carroll MC, Carroll MC. Regulation of B lymphocyte responses to foreign and self-antigens by the CD19/CD21 complex. Annu Rev Immunol. 2000;18(1):393–422.
- Haas KM, Tedder TF. Role of the CD19 and CD21/35 receptor complex in innate immunity, host defense and autoimmunity. Adv Exp Med Biol. 2005;560:125–139.
- Bolland S, Ravetch JV. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity. 2000;13(2):277–285.
- Su K, Yang H, Li X, et al. Expression profile of FcγRIIb on leukocytes and its dysregulation in systemic lupus erythematosus. J Immunol. 2007;178(5):3272–3280.
- Chu SY, Vostiar I, Karki S, et al. Inhibition of B cell receptor-mediated activation of primary human B cells by coengagement of CD19 and FcγRIIb with Fc-engineered antibodies. Mol Immunol. 2008;45(15):3926–3933.
- Szili D, Cserhalmi M, Bankó Z, et al. Suppression of innate and adaptive B cell activation pathways by antibody coengagement of FcγRIIb and CD19. MAbs. 2014;6(4):991–999.
- Makita S, Yoshimura K, Tobinai K. Clinical development of anti-CD19 chimeric antigen receptor T-cell therapy for B-cell non-Hodgkin lymphoma. Cancer Sci. 2017;108(6):1109–1118.
- Ellebrecht CT, Bhoj VG, Nace A, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. 2016;353(6295):179–184.
- Leandro MJ, Cooper N, Cambridge G, Ehrenstein MR, et al. Bone marrow B-lineage cells in patients with rheumatoid arthritis following rituximab therapy. Rheumatology (Oxford). 2007;46(1):29–36.
- Oflazoglu E, Audoly LP. Evolution of anti-CD20 monoclonal antibody therapeutics in oncology. MAbs. 2010;2(1):14–19.
- Weiner GJ. Rituximab: mechanism of action. Semin Hematol. 2010;47(2):115–123.
- Kasi PM, Tawbi HA, Oddis CV, et al. Clinical review: serious adverse events associated with the use of rituximab – a critical care perspective. Crit Care. 2012;16(4):231.
- Weidenbusch M, Rommele C, Schrottle A, et al. Beyond the LUNAR trial. Efficacy of rituximab in refractory lupus nephritis. Nephrol Dial Transplant. 2013;28(1):106–111.
- Terrier B, Amoura Z, Ravaud P, et al. Safety and efficacy of rituximab in systemic lupus erythematosus: results from 136 patients from the French AutoImmunity and Rituximab registry. Arthritis Rheum. 2010;62(8):2458–2466.
- Diaz-Lagares C, Croca S, Sangle S, et al. Efficacy of rituximab in 164 patients with biopsy-proven lupus nephritis: pooled data from European cohorts. Autoimmun Rev. 2012;11(5):357–364.
- Witt M, Grunke M, Proft F, et al. Clinical outcomes and safety of rituximab treatment for patients with systemic lupus erythematosus (SLE) - results from a nationwide cohort in Germany (GRAID). Lupus. 2013;22(11):1142–1149.
- Iaccarino L, Bartoloni E, Carli L, et al. Efficacy and safety of off-label use of rituximab in refractory lupus: data from the Italian Multicentre Registry. Clin Exp Rheumatol. 2015;33(4):449–456.
- Merrill JT, Neuwelt CM, Wallace DJ, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010;62(1):222–233.
- Rovin BH, Furie R, Latinis K, et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum. 2012;64(4):1215–1226.
- Condon MB, Ashby D, Pepper RJ, et al. Prospective observational single-centre cohort study to evaluate the effectiveness of treating lupus nephritis with rituximab and mycophenolate mofetil but no oral steroids. Ann Rheum Dis. 2013;72(8):1280–1286.
- Albert D, Dunham J, Khan S, et al. Variability in the biological response to anti-CD20 B cell depletion in systemic lupus erythaematosus. Ann Rheum Dis. 2008;67(12):1724–1731.
- Ehrenstein MR, Wing C. The BAFFling effects of rituximab in lupus: danger ahead? Nat Rev Rheumatol. 2016;12(6):367–372.
- Sorensen PS, Blinkenberg M. The potential role for ocrelizumab in the treatment of multiple sclerosis: current evidence and future prospects. Ther Adv Neurol Disord. 2016;9(1):44–52.
- Mysler EF, Spindler AJ, Guzman R, et al. Efficacy and safety of ocrelizumab in active proliferative lupus nephritis: results from a randomized, double-blind, phase III study. Arthritis Rheum. 2013;65(9):2368–2379.
- Bologna L, Gotti E, Manganini M, et al. Mechanism of action of type II, glycoengineered, anti-CD20 monoclonal antibody GA101 in B-chronic lymphocytic leukemia whole blood assays in comparison with rituximab and alemtuzumab. J Immunol. 2011;186(6):3762–3769.
- Golay J, Da Roit F, Bologna L, et al. Glycoengineered CD20 antibody obinutuzumab activates neutrophils and mediates phagocytosis through CD16B more efficiently than rituximab. Blood. 2013;122(20):3482–3491.
- Evans SS, Clemmons AB. Obinutuzumab: a novel anti-CD20 monoclonal antibody for chronic lymphocytic leukemia. J Adv Pract Oncol. 2015;6(4):370–374.
- Reddy V, Klein C, Isenberg DA, et al. Obinutuzumab induces superior B-cell cytotoxicity to rituximab in rheumatoid arthritis and systemic lupus erythematosus patient samples. Rheumatology (Oxford). 2017;56(7):1227–1237.
- Thornton CC, Ambrose N, Ioannou Y. Ofatumumab: a novel treatment for severe systemic lupus erythematosus. Rheumatology (Oxford). 2015;54(3):559–560.
- Haarhaus ML, Svenungsson E, Gunnarsson I. Ofatumumab treatment in lupus nephritis patients. Clin Kidney J. 2016;9(4):552–555.
- Lin TS. Ofatumumab: a novel monoclonal anti-CD20 antibody. Pharmgenomics Pers Med. 2010;3:51–59.
- Taylor PC, Quattrocchi E, Mallett S, et al. Ofatumumab, a fully human anti-CD20 monoclonal antibody, in biological-naive, rheumatoid arthritis patients with an inadequate response to methotrexate: a randomised, double-blind, placebo-controlled clinical trial. Ann Rheum Dis. 2011;70(12):2119–2125.
- Sorensen PS, Lisby S, Grove R, et al. Safety and efficacy of ofatumumab in relapsing-remitting multiple sclerosis: a phase 2 study. Neurology. 2014;82(7):573–581.
- Bar-Or A, Grove RA, Austin DJ, et al. Subcutaneous ofatumumab in patients with relapsing-remitting multiple sclerosis: the MIRROR study. Neurology. 2018;90(20):e1805–e14.
- Pillai S, Netravali IA, Cariappa A, et al. Siglecs and immune regulation. Annu Rev Immunol. 2012;30(1):357–392.
- Daridon C, Blassfeld D, Reiter K, et al. Epratuzumab targeting of CD22 affects adhesion molecule expression and migration of B-cells in systemic lupus erythematosus. Arthritis Res Ther. 2010;12(6):R204.
- Dörner T, Shock A, Goldenberg DM, et al. The mechanistic impact of CD22 engagement with epratuzumab on B cell function: implications for the treatment of systemic lupus erythematosus. Autoimmun Rev. 2015;14(12):1079–1086.
- Dörner T, Kaufmann J, Wegener WA, et al. Initial clinical trial of epratuzumab (humanized anti-CD22 antibody) for immunotherapy of systemic lupus erythematosus. Arthritis Res Ther. 2006;8(3):R74.
- Wallace DJ, Kalunian K, Petri MA, et al. Efficacy and safety of epratuzumab in patients with moderate/severe active systemic lupus erythematosus: results from EMBLEM, a phase IIb, randomised, double-blind, placebo-controlled, multicentre study. Ann Rheum Dis. 2014;73(1):183–190.
- Wallace DJ, Hobbs K, Clowse ME, et al. Long-term safety and efficacy of epratuzumab in the treatment of moderate-to-severe systemic lupus erythematosus: results from an open-label extension study. Arthritis Care Res (Hoboken). 2016;68(4):534–543.
- Clowse ME, Wallace DJ, Furie RA, et al. Efficacy and safety of epratuzumab in moderately to severely active systemic lupus erythematosus: results from two phase III randomized, double-blind, placebo-controlled trials. Arthritis Rheumatol. 2017;69(2):362–375.
- Vincent FB, Saulep-Easton D, Figgett WA, et al. The BAFF/APRIL system: emerging functions beyond B cell biology and autoimmunity. Cytokine Growth Factor Rev. 2013;24(3):203–215.
- Nardelli B, Belvedere O, Roschke V, et al. Synthesis and release of B-lymphocyte stimulator from myeloid cells. Blood. 2001;97(1):198–204.
- Bossen C, Tardivel A, Willen L, et al. Mutation of the BAFF furin cleavage site impairs B-cell homeostasis and antibody responses. Eur J Immunol. 2011;41(3):787–797.
- López-Fraga M, Fernández R, Albar J, et al. Biologically active APRIL is secreted following intracellular processing in the Golgi apparatus by furin convertase. EMBO Rep. 2001;2(10):945–951.
- Nakayamada S, Tanaka Y. BAFF- and APRIL-targeted therapy in systemic autoimmune diseases. Inflamm Regen. 2016;36(1):6.
- Treml JF, Hao Y, Stadanlick JE, et al. The BLyS family: toward a molecular understanding of B cell homeostasis. Cell Biochem Biophys. 2009;53(1):1–16.
- Coquery CM, Loo WM, Wade NS, et al. BAFF regulates follicular helper t cells and affects their accumulation and interferon-gamma production in autoimmunity. Arthritis Rheumatol. 2015;67(3):773–784.
- Jacob CO, Pricop L, Putterman C, et al. Paucity of clinical disease despite serological autoimmunity and kidney pathology in lupus-prone New Zealand mixed 2328 mice deficient in BAFF. J Immunol. 2006;177(4):2671–2680.
- Salazar-Camarena DC, Ortiz-Lazareno PC, Cruz A, et al. Association of BAFF, APRIL serum levels, BAFF-R, TACI and BCMA expression on peripheral B-cell subsets with clinical manifestations in systemic lupus erythematosus. Lupus. 2016;25(6):582–592.
- Zollars E, Bienkowska J, Czerkowicz J, et al. BAFF (B cell activating factor) transcript level in peripheral blood of patients with SLE is associated with same-day disease activity as well as global activity over the next year. Lupus Sci Med. 2015;2:(1):e000063.
- Kowalczyk-Quintas C, Chevalley D, Willen L, et al. Inhibition of membrane-bound BAFF by the anti-BAFF antibody belimumab. Front Immunol. 2018;9:2698.
- Furie R, Petri M, Zamani O, et al. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 2011;63(12):3918–3930.
- van Vollenhoven RF, Petri MA, Cervera R, et al. Belimumab in the treatment of systemic lupus erythematosus: high disease activity predictors of response. Ann Rheum Dis. 2012;71(8):1343–1349.
- Manzi S, Sanchez-Guerrero J, Merrill JT, et al. Effects of belimumab, a B lymphocyte stimulator-specific inhibitor, on disease activity across multiple organ domains in patients with systemic lupus erythematosus: combined results from two phase III trials. Ann Rheum Dis. 2012;71(11):1833–1838.
- Dooley MA, Houssiau F, Aranow C, et al. Effect of belimumab treatment on renal outcomes: results from the phase 3 belimumab clinical trials in patients with SLE. Lupus. 2013;22(1):63–72.
- Wallace DJ, Navarra S, Petri MA, et al. Safety profile of belimumab: pooled data from placebo-controlled phase 2 and 3 studies in patients with systemic lupus erythematosus. Lupus. 2013;22(2):144–154.
- Zhang F, Bae S-C, Bass D, et al. A pivotal phase III, randomised, placebo-controlled study of belimumab in patients with systemic lupus erythematosus located in China, Japan and South Korea. Ann Rheum Dis. 2018;77(3):355–363.
- D’Cruz D, Maksimowicz-McKinnon K, Oates J, et al. 200 Efficacy and safety of belimumab in patients of black race with systemic lupus erythematosus: results from the EMBRACE study. Lupus Sci Med. 2019;6(Suppl 1):A149–A50.
- Carter LM, Isenberg DA, Ehrenstein MR. Elevated serum BAFF levels are associated with rising anti-double-stranded DNA antibody levels and disease flare following B cell depletion therapy in systemic lupus erythematosus. Arthritis Rheum. 2013;65(10):2672–2679.
- Manetta J, Bina H, Ryan P, et al. Generation and characterization of tabalumab, a human monoclonal antibody that neutralizes both soluble and membrane-bound B-cell activating factor. J Inflamm Res. 2014;7:121–131.
- Isenberg DA, Petri M, Kalunian K, et al. Efficacy and safety of subcutaneous tabalumab in patients with systemic lupus erythematosus: results from ILLUMINATE-1, a 52-week, phase III, multicentre, randomised, double-blind, placebo-controlled study. Ann Rheum Dis. 2016;75(2):323–331.
- Merrill JT, van Vollenhoven RF, Buyon JP, et al. Efficacy and safety of subcutaneous tabalumab, a monoclonal antibody to B-cell activating factor, in patients with systemic lupus erythematosus: results from ILLUMINATE-2, a 52-week, phase III, multicentre, randomised, double-blind, placebo-controlled study. Ann Rheum Dis. 2016;75(2):332–340.
- Hsu H, Khare SD, Lee F, et al. A novel modality of BAFF-specific inhibitor AMG623 peptibody reduces B-cell number and improves outcomes in murine models of autoimmune disease. Clin Exp Rheumatol. 2012;30(2):197–201.
- Furie RA, Leon G, Thomas M, et al. A phase 2, randomised, placebo-controlled clinical trial of blisibimod, an inhibitor of B cell activating factor, in patients with moderate-to-severe systemic lupus erythematosus, the PEARL-SC study. Ann Rheum Dis. 2015;74(9):1667–1675.
- Merrill JT, Shanahan WR, Scheinberg M, et al. Phase III trial results with blisibimod, a selective inhibitor of B-cell activating factor, in subjects with systemic lupus erythematosus (SLE): results from a randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. 2018;77(6):883–889.
- Richard AF, Thomas M, Chu A, et al. Effects of chronic treatment with blisibimod, an inhibitor of B cell activating factor, on renal and inflammation biomarkers in patients with systemic lupus erythematosus: observations from the placebo-controlled pearl-SC and open-label extension studies. 2013 ACR/ARHP Annual Meeting. [cited 2018 Dec 10]. Available from: https://acrabstracts.org/abstract/
- Dall'Era M, Chakravarty E, Wallace D, et al. Reduced B lymphocyte and immunoglobulin levels after atacicept treatment in patients with systemic lupus erythematosus: results of a multicenter, phase Ib, double-blind, placebo-controlled, dose-escalating trial. Arthritis Rheum. 2007;56(12):4142–4150.
- Ginzler EM, Wax S, Rajeswaran A, et al. Atacicept in combination with MMF and corticosteroids in lupus nephritis: results of a prematurely terminated trial. Arthritis Res Ther. 2012;14(1):R33.
- Merrill JT, Wallace DJ, Wax S, et al. Efficacy and safety of atacicept in patients with systemic lupus erythematosus. Arthritis Rheumatol. 2018;70(2):266–276.
- Wallace D, Isenberg D, Kao A, et al. S7A:4 Reduction of systemic lupus flares by atacicept in a randomised, placebo-controlled, phase iib study (address ii) and its extension study. Lupus Science & Medicine. 2018;5(Suppl 1):A25–A6.
- Tackey E, Lipsky PE, Illei GG. Rationale for interleukin-6 blockade in systemic lupus erythematosus. Lupus. 2004;13(5):339–343.
- Solus JF, Chung CP, Oeser A, et al. Genetics of serum concentration of IL-6 and TNFalpha in systemic lupus erythematosus and rheumatoid arthritis: a candidate gene analysis. Clin Rheumatol. 2015;34(8):1375–1382.
- da Silva JC, Mariz HA, da Rocha Júnior LF, et al. Hydroxychloroquine decreases Th17-related cytokines in systemic lupus erythematosus and rheumatoid arthritis patients. Clinics (Sao Paulo). 2013;68(6):766–771.
- Liang B, Gardner DB, Griswold DE, et al. Anti-interleukin-6 monoclonal antibody inhibits autoimmune responses in a murine model of systemic lupus erythematosus. Immunology. 2006;119(3):296–305.
- Rovin BH, van Vollenhoven RF, Aranow C, et al. A multicenter, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of treatment with sirukumab (CNTO 136) in patients with active lupus nephritis. Arthritis Rheumatol. 2016;68(9):2174–2183.
- Ablynx Vobarilizumab fails in lupus trial. Drug updated information. [cited 2019 Jul]. Available from: http://www.pharmatimes.com/news/ablynx_vobarilizumab_fails_in_lupus_trial_1229404
- Crow MK. Type I interferon in the pathogenesis of lupus. J Immunol. 2014;192(12):5459–5468.
- Hanten JA, Vasilakos JP, Riter CL, et al. Comparison of human B cell activation by TLR7 and TLR9 agonists. BMC Immunol. 2008;9(1):39.
- Ittah M, Miceli-Richard C, Eric Gottenberg J, et al. B cell-activating factor of the tumor necrosis factor family (BAFF) is expressed under stimulation by interferon in salivary gland epithelial cells in primary Sjögren's syndrome. Arthritis Res Ther. 2006;8(2):R51.
- Farkas L, Beiske K, Lund-Johansen F, et al. Plasmacytoid dendritic cells (natural interferon- alpha/beta-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am J Pathol. 2001;159(1):237–243.
- Kirou KA, Lee C, George S, et al. Activation of the interferon-α pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum. 2005;52(5):1491–1503.
- Niewold TB, Swedler WI. Systemic lupus erythematosus arising during interferon-alpha therapy for cryoglobulinemic vasculitis associated with hepatitis C. Clin Rheumatol. 2005;24(2):178–181.
- Kalunian KC, Merrill JT, Maciuca R, et al. A phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon-alpha) in patients with systemic lupus erythematosus (ROSE). Ann Rheum Dis. 2016;75(1):196–202.
- Yao Y, Richman L, Higgs BW, et al. Neutralization of interferon-α/β–inducible genes and downstream effect in a phase I trial of an anti–interferon-α monoclonal antibody in systemic lupus erythematosus. Arthritis Rheum. 2009;60(6):1785–1796.
- Petri M, Wallace DJ, Spindler A, et al. Sifalimumab, a human anti–interferon-α monoclonal antibody, in systemic lupus erythematosus: a phase I randomized, controlled, dose-escalation study. Arthritis Rheum. 2013;65(4):1011–1021.
- Khamashta M, Merrill JT, Werth VP, et al. Sifalimumab, an anti-interferon-alpha monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double-blind, placebo-controlled study. Ann Rheum Dis. 2016;75(11):1909–1916.
- Peng L, Oganesyan V, Wu H, et al. Molecular basis for antagonistic activity of anifrolumab, an anti-interferon–α receptor 1 antibody. MAbs. 2015;7(2):428–439.
- Riggs JM, Hanna RN, Rajan B, et al. Characterisation of anifrolumab, a fully human anti-interferon receptor antagonist antibody for the treatment of systemic lupus erythematosus. Lupus Sci Med. 2018;5(1):e000261.
- Clark LB, Foy TM, Noelle RJ. CD40 and its ligand. Adv Immunol. 1996;63:43–78.
- Elgueta R, Benson MJ, de Vries VC, et al. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. 2009;229(1):152–172.
- Strom L, Laurencikiene J, Miskiniene A, et al. Characterization of CD40-dependent immunoglobulin class switching. Scand J Immunol. 1999;49(5):523–532.
- Schonbeck U, Libby P. The CD40/CD154 receptor/ligand dyad. Cmls, Cell Mol Life Sci. 2001;58(1):4–43.
- Aloui C, Prigent A, Sut C, et al. The signaling role of CD40 ligand in platelet biology and in platelet component transfusion. IJMS. 2014;15(12):22342–22364.
- Lu Q, Wu A, Tesmer L, et al. Demethylation of CD40LG on the inactive X in T cells from women with lupus. J Immunol. 2007;179(9):6352–6358.
- Zhou Y, Yuan J, Pan Y, et al. T cell CD40LG gene expression and the production of IgG by autologous B cells in systemic lupus erythematosus. Clin Immunol. 2009;132(3):362–370.
- Boumpas DT, Furie R, Manzi S, et al. A short course of BG9588 (anti–CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum. 2003;48(3):719–727.
- Sidiropoulos PI, Boumpas DT. Lessons learned from anti-CD40L treatment in systemic lupus erythematosus patients. Lupus. 2004;13(5):391–397.
- Tocoian A, Buchan P, Kirby H, et al. First-in-human trial of the safety, pharmacokinetics and immunogenicity of a PEGylated anti-CD40L antibody fragment (CDP7657) in healthy individuals and patients with systemic lupus erythematosus. Lupus. 2015;24(10):1045–1056.
- Satterthwaite AB. Bruton’s Tyrosine Kinase, a component of B cell signaling pathways, has multiple roles in the pathogenesis of lupus. Front Immunol. 2018;8:1986.
- Haselmayer P, Camps M, Liu-Bujalski L, et al. Efficacy and pharmacodynamic modeling of the BTK inhibitor evobrutinib in autoimmune disease models. J Immunol. 2019;202(10):2888–2906.
- Chalmers SA, Wen J, Doerner JL, et al. BTK inhibition ameliorates renal, skin, and brain disease in a spontaneous murine model of systemic lupus erythematosus. J Immunol. 2018;200(1):175.