
Abstract
Autoimmune gastrointestinal dysmotility (AGID), an idiopathic or paraneoplastic phenomenon, is a clinical form of limited autoimmune dysautonomia. The symptoms of AGID and gastrointestinal manifestations in patients with autoimmune rheumatic diseases are overlapping. Antineuronal autoantibodies are often detected in patients with AGID. Autoantibodies play a key role in GI dysmotility; however, whether they cause neuronal destruction is unknown. Hence, the connection between the presence of these autoantibodies and the specific interference in synaptic transmission in the plexus ganglia of the enteric nervous system has to be determined. The treatment options for AGID are not well-defined. However, theoretically, immunomodulatory therapies have been shown to be effective and are therefore used as the first line of treatment. Nonetheless, diverse combined immunomodulatory therapies should be considered for intractable cases of AGID. We recommend comprehensive autoimmune evaluation and cancer screening for clinical diagnosis of AGID. Univocal diagnostic criteria, treatment protocols, and outcome definitions for AGID are required for prompt diagnosis and treatment and appropriate management of immunotherapy, which will circumvent the need for surgeries and improve patient outcome. In conclusion, AGID, a disease at the interface of clinical immunology and neurogastroenterology, requires further investigations and warrants cooperation among specialists, especially clinical immunologists, gastroenterologists, and neurologists.
1. Introduction
Gastrointestinal (GI) motility disorder is observed in a large variety of diseases (). Autoimmune GI dysmotility (AGID) is recognized as a limited form of autoimmune dysautonomia [Citation1]. One of the earliest descriptions of patients who had severe GI dysmotility with small-cell lung carcinoma (SCLC) was by Lennon et al. in 1991 [Citation2]. They detected IgG antibodies that were reactive with neurons of the myenteric and submucosal plexuses of jejunum and stomach in 4 of 5 patients with small-cell carcinoma; all 5 patients had chronic intestinal pseudo-obstruction (IPO). Sheikh and Shaw–Stiffel reported the GI manifestations of Sjögren's syndrome in 1995 [Citation3], thereafter, it became known that GI dysmotility occurs frequently in autoimmune rheumatic diseases. AGID is a relatively recently discovered neurogastroenterological disorder. Since the 2000s, AGID has been proposed to be an immune-mediated autonomic dysfunction and often a paraneoplastic syndrome [Citation1,Citation4]. However, currently, no criteria or consensus for early diagnosis and management of AGID is available. Although AGID is currently diagnosed on the basis of the detection of the autoantibody (AAb) and/or the history of recent or past neoplasia, it is unknown whether they indeed cause neurological defects. AGID warrants exclusion diagnosis in the clinical context.
Table 1. Gastrointestinal motility disorders.
The present review analyzes and summarizes the current status of basic and clinical research on AGID. The order of appearance of concepts discussed in the review is as follows: actual perspectives, the anatomical stricture of the enteric nervous system, epidemiology and clinical features, pathophysiology, therapy and therapeutic targets, and future perspectives. Ultimately, we have attempted to formulate a potential procedure for the diagnosis and treatment of AGID based on the literature.
2. Actual perspectives on AGID
According to previous reports, achalasia, gastroparesis, pyloric stenosis, IPO, slow intestinal transit, colonic inertia, and anal spasm are considered to represent AGID () [Citation1,Citation4]. However, immune-mediated mechanisms are not responsible for all the above diseases. The etiology of these disorders includes autoimmune, infectious, inflammatory, degenerative, and familial causes. Clinical presentations include dysphagia, odynophagia, postprandial epigastric discomfort, early satiety, nausea, vomiting, bloating, involuntary weight loss with malnutrition, abdominal pain, and intractable constipation or diarrhea. Subtle or overt neurological manifestations, especially autonomic symptoms, may simultaneously accompany the GI manifestation [Citation1,Citation4,Citation5]. AGID was often confirmed in patients with autoimmune autonomic anglionopathy (AAG). AAG, an acquired immune-mediated disorder that leads to autonomic failure, is mediated by autoantibodies targeting the ganglionic nicotinic acetylcholine receptor (gAChR) [Citation6–8]. Patients with AAG showed impaired sympathetic (orthostatic hypotension, and anhidrosis) and parasympathetic symptoms (abnormal pupillary response, sexual dysfunction, and fixed heart rate). Recently, we demonstrated the prevalence of anti-gAChR AAbs in patients with achalasia and chronic IPO in Japan [Citation5]. We speculate that anti-gAChR AAbs might mediate autonomic dysfunction, contributing to autoimmune mechanisms underlying these GI dysmotilities.
Figure 1. Autoimmune gastrointestinal dysmotility (AGID). AGID and the corresponding parts affected in the GI tract have been listed.
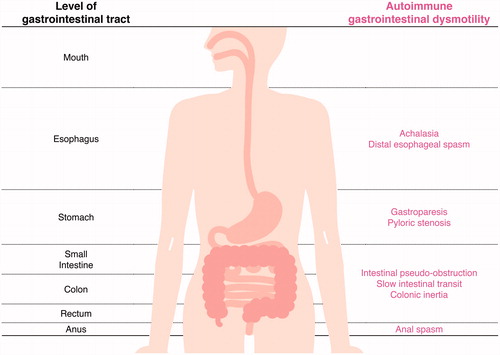
However, the clinical application of AAb measurement is limited. In fact, AAb measurement is not routinely performed and remains limited to patients with uncertain diagnosis. AGID is actually classified according to the anatomical position and etiology. AGID can occur at every level of the alimentary tract, i.e. esophagus, stomach, small intestine, colon, rectum and anus. Based on its etiology, Flanagan et al. suggested a comprehensive neural AAb testing system, in which they evaluated immunotherapy as a method for diagnosis and treatment at the Mayo Clinic [Citation4]. At that time, they proposed the inclusion criteria of AGID. An autoimmune etiology is assumed for AGID cases seropositive for any of the AAbs. However, paraneoplastic AGID is considered to be triggered by the presence of tumors, even if the sera of these patients are AAb-positive.
3. The enteric nervous system
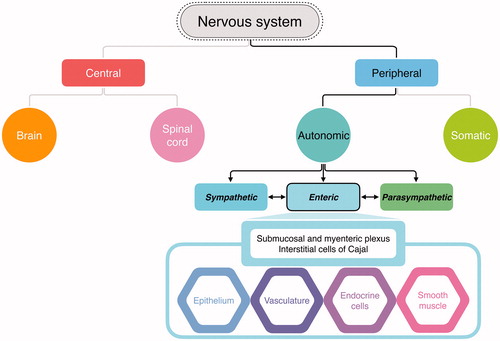
The human nervous system controls voluntary and involuntary actions via the central nervous system (CNS) and the peripheral nervous system (PNS). The CNS consists of the brain and the spinal cord and acts as the main control center. The PNS consists of the associated nerve networks, which connect parts of the body to the CNS. The PNS is divided into the autonomic nervous system, which controls involuntary activities such as digestion and breathing, and the somatic nervous system, which controls voluntary actions by transmitting stimuli information to the CNS and sending back response signals to striated muscles. The autonomic nervous system is further divided into the sympathetic, parasympathetic, and enteric nervous system (ENS). The sympathetic nervous system deals with fight-or-flight signals, the parasympathetic system maintains body conditions, and the enteric nervous system regulates GI functions. The ENS is a large division of the PNS that can control GI behavior independent of CNS input (). The ENS may affect the effector systems in the gut directly or indirectly via its action on intermediate cells, which include the epithelium, smooth muscles, blood vessels, endocrine cells, and the interstitial cells of Cajal [Citation9].
Figure 2. Division and function of the nervous system. The autonomic nervous system is a part of the peripheral nervous system that acts as an involuntary control system below the level of consciousness and controls visceral functions. The enteric nervous system (ENS) is a division of autonomic nervous system that regulates the gastrointestinal (GI) system. While the ENS communicates with the central nervous system (CNS) and regulates digestive functions, the ENS can also control local enteric reflexes independent of the CNS. The ENS can detect and integrate mechanical or chemical changes in the GI tract, thereby signaling effector cells, including the epithelium, smooth muscle, vessels, and endocrine cells, and assists in maintaining GI homeostasis.
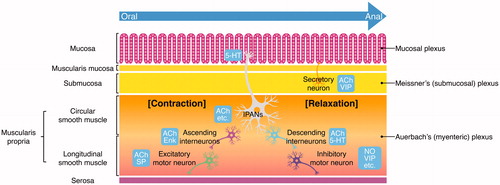
The ENS performs multiple roles: determining the patterns of movement of the GI tract, controlling gastric acid secretion, regulating movement of fluid across the epithelial lining, changing local blood flow, modifying nutrient handling, and interacting with the immune and endocrine systems of the gut [Citation10]. The GI tract is innervated by intrinsic neurons belonging to the ENS and by the axons of extrinsic sympathetic, parasympathetic, and visceral afferent neurons [Citation11,Citation12]. For the intrinsic innervation of the GI, the ENS is composed mainly of two ganglionated plexuses that form a network of neurons and glial cells (): an outer plexus lying between the longitudinal and circular muscle layers, called the myenteric plexus or Auerbach’s plexus, and an inner plexus, called the submucosal plexus or Meissner’s plexus, which lies in the submucosa [Citation10–12]. The nerve connections within and between these two plexuses are also shown in . Enteric neurons include intrinsic primary afferent neurons (IPANs), interneurons, motor neurons, secretomotor neurons, and vasomotor neurons. These ENS neurons are synaptically interconnected to form parallel reflex circuits [Citation10–13]. The neurochemical signaling within the ENS is extremely complex. In an attempt to improve the understanding regarding the multiple functions of the ENS, some research groups have identified neurotransmitters that are released by the nerve endings of different types of enteric neurons. Acetylcholine (ACh) mostly stimulates GI activity such as up-regulation of Cl- current across the epithelial lining and of GI smooth muscle contractility. The other major transmitters include nitric oxide (NO), vasoactive intestinal polypeptide (VIP), substance P, neuropeptide Y (NPY), and adenosine-triphosphate (ATP) [Citation10–12]. The specific functions of many of these are becoming clear.
Figure 3. The organization of the ENS within the intestinal wall. Local distension of the intestinal wall, distortion of the mucosa, and chemical contents in the lumen activate the intrinsic primary afferent neurons (IPANs) located in both the submucosal plexus and myenteric plexus. IPANs activate ascending and descending interneurons, which stimulate excitatory and inhibitory motor neurons, respectively. ACh refers to neurons that contain acetylcholine. SP refers to neurons that contain substance P. Enk refers to encephalin-expressing ascending interneurons. NO and VIP indicate inhibitory motor neurons that secrete nitric oxide and vasoactive intestinal peptide, respectively. Secretomotor and vasomotor neurons of the submucosal plexus secrete ACh or VIP.
4. Epidemiology and clinical features of AGID
The epidemiological data of AGID are not conclusive and the incidence of AGID has not yet been estimated. The first comprehensive study by Dhamija et al. conducted at Mayo Clinic reported the clinical profiles of 24 patients with severe GI dysmotility [Citation1]. These 24 patients were serologically ascertained as having one or more AAbs consistent with neurologic autoimmunity in the course of service serologic evaluation in the Mayo Neuroimmunology Laboratory. Sixteen of the 24 patients of this report were female (67%) and the median age was 59 years. GI dysmotility included esophageal dysmotility in 8 (6 had achalasia), delayed gastric emptying in 12, slow small intestinal transit in 7, slow colonic transit in 4, and pelvic floor dyssynergia in 4 patients. Four patients underwent abdominal surgery and 2 were put on total parenteral nutrition. The following cation channel AAbs were detected in 23 patients: neuronal voltage-gated calcium channel (VGCC; 6 patients), acetylcholine receptor (11 patients with ganglionic-type and 4 with muscle-type), and neuronal voltage-gated potassium channel AAbs (VGKC; 4 patients). Two patients had type 1 antineuronal nuclear AAbs. Approximately half of the patients had neural AAbs or other antibody markers of organ-specific autoimmunity (thyroid- or gastric parietal cell-specific). Neoplasia in the lung, breast, endometrial lining, GI, and thymus (thymoma) was diagnosed in 11 patients.
Flanagan et al. at Mayo Clinic consecutively evaluated 23 of 39 patients with AGID (median of 5 patients/year from August 2006 to February 2014) who fulfilled their inclusion criteria [Citation4]: (1) Prominent symptoms of GI dysmotility with confirmatory baseline scintigraphic evidence of GI hypomotility (i.e. slow gastric emptying, delayed transit through small intestine or large intestine), abnormal manometry findings or symptoms of severe GI dysmotility with evidence of pandysautonomia. (2) An autoimmune etiology suspected based on clinical suspicion (subacute onset), seropositivity for a neural AAb, or personal/family history of autoimmune disease. (3) Treated by immunotherapy (without changes in other medications) for 4–12 weeks on a trial basis (intravenous high-dose immunoglobulin or methylprednisolone). Twenty-one of the 23 patients (91%) were female, and the median age at the time of treatment was 38 years. They checked the personal and family history of autoimmunity, serological evidence of extra neurological autoimmunity, and the history of recent or past neoplasia or risk factors for cancer. Furthermore, they performed GI transit testing using scintigraphy, manometry studies for gastroduodenum and colon, autonomic testing, and neural autoantibody evaluation including AAbs specific for neuronal nuclear antigens, neuronal cytoplasmic antigens, cation channels (neuronal VGCC, VGKC complex, nicotinic AChRs extracted from ganglionic neurons and muscle), muscle striational antigens, and recombinant GAD. The clinical characteristics of the 17 responders to immunotherapies were tabulated. Fifteen of the 17 responders reported constipation, 13 reported nausea/vomiting, 12 had severe weight loss, and 9 complained of bloating. Extra-intestinal autonomic symptoms included orthostasis in 10 cases and visual accommodation problems in 4 cases. Autonomic dysfunction was broadly classified as pandysautonomia (all divisions of the autonomic nervous system affected; 4 cases), multifocal dysautonomia (multiple divisions of the autonomic system affected; 7 cases), and limited dysautonomia (autonomic dysfunction restricted to the GI system; 6 cases). Ten patients (59%) had coexisting autoimmune disorders (Graves’ disease, celiac disease, Hashimoto thyroiditis, lupus, alopecia areata, type 1 diabetes, hypothyroidism, Crohn’s disease, maternal hypothyroidism, and maternal diabetes). Serological evaluation revealed a neural-specific AAb in 12 of 17 responders (71%). Specifically, AAbs were detected against gAChR in 6 cases, ANNA-1 in 3 cases, VGKC-complex in 3 cases, striated muscle in 3 cases, peripherin in 2 cases, VGCC N-type in 1 case, and GAD65 in 1 case. Seven of sixteen patients (44%) were seropositive for the antinuclear antibody. Double stranded DNA antibody was detected in 1 of the 2 patients who had systemic lupus erythematosus. Two of 10 patients tested harbored 1 or more antibodies to extractable nuclear antigens (SSA, 2; Sm, 1; ribonucleoprotein, 1). Three patients were seropositive for ANNA-1 and had paraneoplastic AGID. All 3 cases had SCLC, thymoma, and small cell carcinoma of the uterine cervix.
Previously, we have observed that Japanese females are predominantly affected by AAG with severe dysmotility [Citation5]. Six of 123 seropositive AAG patients presented with achalasia or diffuse esophageal spasm as severe upper GI dysmotility in this study. The mean age and mean age at disease onset were 49.7 years and 37.8 years, respectively. A gradual mode of onset was more common, and the GI tract symptoms included various digestive system problems, such as appetite loss, nausea and/or vomiting, early satiety, and postprandial abdominal pain owing to the dysfunction of the upper digestive system, and diarrhea, constipation, alternate stool abnormality, and paralytic ileus owing to the dysfunction of the lower digestive system. Among the 123 patients with seropositive AAG, 4 patients had paralytic ileus, one of whom disclosed having gastroparesis as severe lower GI dysmotility. The mean age and mean age at disease onset were 63.5 years and 60.5 years, respectively. A gradual mode of onset was frequently observed and widespread dysfunction of the digestive system was common. Other autonomic symptoms (orthostatic hypotension/intolerance and bladder dysfunction) and extra-autonomic manifestations were observed in these 10 patients. In particular, regarding extra-autonomic manifestations, autoimmune diseases (Sjögren’s syndrome and primary biliary cirrhosis) were detected in 2 patients and gastric cancer was detected in 1 patient. Furthermore, we attempted to clarify the seroprevalence of gAChR AAbs in the patients with severe GI dysmotility in this previous study. We enrolled 28 patients with achalasia and 14 patients with IPO and detected anti-gAChR AAbs in 21.4% patients with achalasia and in 50.0% patients with chronic IPO. Although the patients with achalasia and chronic IPO demonstrated various autonomic dysfunction, bladder dysfunction was observed in the seropositive patients with chronic IPO as a prominent clinical characteristic of dysautonomia. The seropositive IPO group showed female predominance (71%). Two of seven cases of seropositive IPO had Sjögren’s syndrome and cancer of the uterine body. However, in our study, no significant difference was noted in the coexistence of autoimmune diseases and tumors between the seropositive and seronegative IPO groups.
In summary, as AGID is assumed to be a rare disease, we highly recommend comprehensive autoimmune serological evaluation and neoplasm screening for diagnosing AGID. This will enable us to determine the true incidence rate of AGID and establish the clinical criteria for AGID.
5. Pathophysiology of AGID: autoimmunity and GI dysmotility
5.1. Heterogeneity of AGID
AGID is a clinically heterogeneous group of disorders in which symptoms are presumed to arise as a result of the dysfunction of the ENS. Several immune mechanisms have been proposed to contribute to the pathophysiology of a range of gut motility disorders. AAbs specific for neuronal antigens, which are known to be pathogenic in some peripheral neuromuscular diseases, are present in some individuals with GI dysmotility [Citation14]. Circulating AAbs targeting the ENS and leading to disrupted motility have been demonstrated in paraneoplastic syndromes [Citation15], Lambert Eaton syndrome [Citation16], Chagas disease [Citation17,Citation18], diabetes [Citation16,Citation19], achalasia [Citation20–25], and IPO [Citation2,Citation26–29]. This is best understood in paraneoplastic cases of GI dysmotility, where antibodies against neuronal antigens expressed by tumor cells cross-react with native neurons leading to dysfunction [Citation1,Citation14,Citation28]. Antineuronal AAbs have also been found in AGID affecting every level of the gut () [Citation1,Citation5,Citation6,Citation22,Citation30–34].
Table 2. Antineuronal autoantibodies detected in patients with gastrointestinal dysmotility.
5.2. Humoral immunity in AGID
Among these antineuronal AAbs, autoimmunity to the anti-gAChR AAbs has been investigated so far [Citation8,Citation35,Citation36]. As mentioned previously, we attempted to determine the associations between autonomic dysfunction, anti-gAChR AAbs, and clinical features in patients with GI motility disorders, including those with achalasia and chronic IPO. In the peripheral autonomic ganglia, neuronal AChRs (=gAChR) are expressed by neurons in sympathetic, parasympathetic, and enteric ganglia. Patients with AAG often harbor AAbs against gAChR, which may disrupt synaptic transmission in autonomic ganglia and lead to autonomic failure. The anti-gAChR AAbs might mediate autonomic dysfunction, contributing to the autoimmune mechanisms underlying GI motility disorders. We focused on autonomic dysfunction in autoimmune rheumatic diseases (ARD), including Sjögren’s syndrome, systemic sclerosis (SSc), rheumatoid arthritis, and systemic lupus erythematosus [Citation27,Citation29,Citation37–42]. GI disorders have been reported in a wide variety of systemic autoimmune diseases. Recently, we attempted to understand the pathogenesis of GI manifestations in SSc, as it is well known that SSc frequently affects the GI functions [Citation43]. The patients with SSc often suffer from esophageal absent contractility, severe gastroesophageal reflux disease, IPO and malabsorption with the complaints of dysphasia, heartburn, vomiting, abdominal fullness, constipation and diarrhea. Previous studies have demonstrated that GI dysmotility in SSc was associated with circulating AAbs against the muscarinic AChRs, RNPC3, U1 snRNP, U3 snRNP, signal recognition particle, Ku, and myenteric neurons [Citation40,Citation44–52]. We have reported the clinical characteristics of SSc patients with seropositivity for the anti-gAChR AAbs, and have analyzed the relationship among gAChR Abs, several biomarkers, and GI manifestations in SSc [Citation43]. In AGID, including ARD-related GI dysmotility, the connection between the presence of these AAbs and the specific interference in synaptic transmission in the plexus ganglia of the ENS should be determined. Other than the gAChR AAbs, Hubball et al. verified the presence and distribution of VGKCs in human ENS, and reported that the VGKC AAbs act pathologically in GI dysmotility. Furthermore, they reported that potassium KV1 channels are abundant throughout the human GI tract and are expressed by enteric neurons of the myenteric and submucosal plexuses in a regionally specific pattern in immunohistochemical staining [Citation14].
5.3. Cellular immunity in AGID including animal model studies
Although the above reports indicate that AAbs play a key role in GI dysmotility, whether they indeed cause neuronal destruction is not known. In patients with post-infectious irritable bowel syndrome, quantitative histology showed evidence of continuing inflammation, with elevated intra-epithelial T cell levels in rectal biopsy [Citation53,Citation54]. Hence, the effect of cellular immune reactions on AGID should be investigated.
Active immunization models of AGID and AAG have been established. Meeusen and colleagues in Mayo Clinic reported that AGID is adequately induced in mice via active immunization of live nicotinic AChRα3-expressing xenogeneic cells [Citation55]. They showed severe hypomotility associated with reduction of enteric nicotinic AChRα3 in spite of not causing neuronal cell loss in the enteric ganglia, and they concluded that ‘self’ nicotinic AChRα3-reactive AAb production is required to develop AGID in actively immunized mice; furthermore, these phenomena in the murine model are indicative of an IgG-mediated, rather than T cell-mediated, pathogenesis. In terms of animal model of AAG, the Mayo group showed that rabbits immunized with a recombinant α3 neuronal nicotinic AChR subunit fusion protein can produce gAChR AAbs and developed autonomic failure [Citation56,Citation57]. Rabbits with high gAChR AAb levels do not gain weight and present with gastroparesis. The severity of autonomic failure correlates with serum antibody levels. Immunohistochemical staining of superior cervical ganglia and myenteric plexus neurons demonstrates intact presynaptic nerve terminals and postsynaptic neurons containing cytoplasmic nAChR with reduced surface nAChR. Hence, they concluded that this animal model of human AAG is a disorder of ganglionic cholinergic synaptic transmission caused by gAChR AAbs. The same research group has successfully established a passive transfer mouse model using rabbit IgG containing gAChR AAbs [Citation58]. Recipient mice developed transient GI dysmotility, indicating that dysautonomia in recipient mice mimicked an antibody-mediated pathogenesis for AAG.
Sanchez-Ruiz et al. reported CD8 T cell-mediated enteric murine ganglionitis [Citation59]. Based on the high prevalence of bowel dysmotility in patients with inflammatory bowel disease, transgenic mice expressing ovalbumin on enteric neurons were generated via induction of severe enteric ganglionitis after adoptive transfer of ovalbumin-specific CD8 T cells. Twenty-four hours after the onset of colitis-related symptoms, 60% of the submucosal and myenteric plexus neurons were lost, resulting in severely impaired GI transition. They further demonstrated that autoimmune CD8 T cells play an important pathogenetic role in GI dysmotility and may destroy enteric neurons. Whether the passive transfer of any of the AAbs from human AGID/AAG patients to experimental animals can reproduce the clinical and laboratory features of the disease should be verified. As pointed out by Drachman, this will confirm the specific pathogenic role of the antibody and will rule out a cell-mediated disease mechanism [Citation60]. GI dysmotility occurs in experimental autoimmune encephalomyelitis (EAE), a murine model of multiple sclerosis (MS) [Citation61,Citation62]. Delayed colonic motility is exhibited in wild type mice induced with EAE but is absent in B cell-deficient mice, suggesting that the observed dysmotility is antibody mediated [Citation61]. Further experiments are required to dissect the complexity, namely the interactions among the ENS cells, immune cells, and humoral factors in animal models of AGID.
5.4. Gut dysbiosis in AGID
The bidirectional brain-gut microbiota interactions are believed to be involved in the pathogenesis of well-known brain-gut disorders such as irritable bowel syndrome (IBS) and other GI disorders [Citation63,Citation64] and have more recently been implicated as a possible mechanism in the pathophysiology of neurological disorders such as Parkinson’s disease [Citation65,Citation66], autism spectrum disorders [Citation67,Citation68]. The next challenge is to elucidate the role of brain-gut axis in AGID, because AGID is a neurological, GI, and an autoimmune disease [Citation69]. With regard to PNS, the vagus nerve, which mainly constitutes the parasympathetic nervous system, sends signals from the brain to visceral organs including the heart, lungs, and intestine via ACh [Citation70]. Tracey and colleagues discovered the cholinergic ‘anti-inflammatory reflex’ as a major neural circuit that modulates immune responses [Citation71,Citation72]. In this reflex, peripheral inflammation is sensed by vagal afferent neurons, activating a brainstem circuit that leads to decreased cytokine production via vagal afferent neuron signaling [Citation70]. Most recently, Teratani et al. reported the biological mechanism by which bacterial information from the intestinal tract is integrated in the liver and transmitted to the brain to control the production of intestinal regulatory T cells through the vagal nerve reflex [Citation73]. Thus, several experimental approaches have been reported on autonomic innervation of immune organs and neuroimmune modulation [Citation74].
5.5. Functional GI disorders or AGID?
We have to mention the functional GI disorders (FGIDs). Epidemiological studies from the UK have suggested that FGIDs are associated with atopy and rheumatological autoimmune diseases [Citation75,Citation76]. Functional dyspepsia (FD) and IBS and are the most common FGIDs affecting 10–20% of the general population in Japan. In a large population-based study in Australia, Koloski et al. demonstrated that food allergy, psoriasis, and rheumatoid arthritis were independent risk factors for IBS, and asthma and food allergy were independent predictors of FD [Citation77]. They indicated that immune-mediated mechanism, i.e. allergic and autoimmune conditions, may be involved in the pathogenesis of certain FGIDs. Are FGIDs another subtype of AGID? The incidence of autoimmune disease or allergic conditions in the patient group of FGIDs in Japan should be surveyed in the near future.
6. Therapy and therapeutic targets in AGID
Controlled treatment trials for AGID are lacking and the optimal therapy remains uncertain; however, clinical observations in individual cases have shown that treatment efficacy varies between individuals. Currently, symptomatic therapies and causal therapies for AGID are available, and symptomatic therapies that are useful for management include the use of antiemetics, prokinetic agents, and cholinesterase inhibitors [Citation4,Citation31].
In theory, immunotherapy should be considered during the acute phase of AGID to inhibit the misregulation of immune response. Intravenous methylprednisolone (IVMP), intravenous immunoglobulin (IVIg), and plasmapheresis (PP) are considered safe and appropriate therapies. Several case reports have demonstrated the effect of these first-line immunotherapies on patients with achalasia or IPO [Citation78–81]. Flanagan et al. assessed a retrospective case series consisting of 23 patients with AGID who completed a 6–12-week therapeutic trial of IVMP and/or IVIg [Citation4]. The condition of 17 patients improved after immunotherapy. Symptomatic improvements correlated with improvement in GI motility and autonomic function in scintigraphic, manometric, and autonomic tests. Based on our experience with immunotherapy of patients with AGID, we recommend using high doses of IVMP, IVIg, or PP [Citation5,Citation32,Citation34]. Similar to the treatment of AAG, combined immunomodulatory therapies using immunosuppressive agents, including combination of orally administered prednisolone with PP or IVIg, were effective for some intractable cases [Citation82–84]. Certain case reports in literature mention different treatment regimens [Citation85–88]. One case report showed success with octreotide treatment [Citation85]. Another reported marginal results with a regimen including prednisone, azathioprine, and budesonide [Citation86]. Two others reported success with B cell depletion therapy using rituximab or rituximab and cyclophosphamide [Citation87,Citation88]. One study reported resolution of gastroparesis after resection of a primary retroperitoneal leiomyosarcoma, which is believed to be caused by a similar autoimmune mechanism [Citation89]. Management of AGID depends on the underlying etiology. Treatment of patients with paraneoplastic AGID includes treatment of tumors when applicable and symptomatic management [Citation90].
Spear and Mawe indicated the key players in dysmotility and ENS-related therapeutic targets [Citation91], which are as follows: (1) changes in serotonin signaling, protective actions of 5-hydroxytryptamine receptor 4 activation, and proinflammatory actions of serotonin; (2) inflammation-induced enteric neuroplasticity; (3) purinergic neuromuscular transmission; (4) antibody-mediated GI dysmotility; (5) muscularis macrophages, which include cholinergic anti-inflammatory pathway and macrophage β2-adrenergic receptors; (6) enteric glia. In addition, they mentioned the use of cyclooxygenase inhibitors, free radical scavengers, and B cell depletion, as well as the cholinergic anti-inflammatory pathway stimulation and 5-HT4 agonist treatment as potential treatment strategies.
7. Concluding remarks and future perspectives
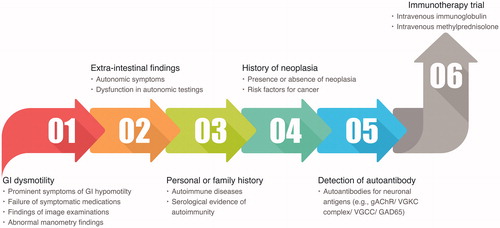
Confusion regarding the definition of AGID and diagnostic/therapeutic protocols renders the evaluation and management of AGID difficult. The chronic IPO is historically misdiagnosed, resulting in unnecessary surgeries, often with dire consequences [Citation92]. Univocal diagnostic criteria, treatment protocols, and outcome definitions are required for prompt diagnosis and treatment and for appropriate management of immunotherapy, which will assist in circumventing the need for unnecessary surgeries and improving patient outcome. Future research, prospective studies, and clinical trials should define the univocal diagnostic criteria for AGID, treatment indications, and therapeutic protocols. A multidisciplinary approach is crucial and may represent the first step for optimizing the therapeutic management of these patients. Moreover, identification of biomarkers that can predict treatment responsiveness and antigens of AGID may be useful for developing targeted and personalized therapies. We have presented the current procedure for diagnosis and treatment of AGID based on previous studies [Citation1,Citation4,Citation5] ().
Figure 4. Current procedure for diagnosis and treatment of AGID. We recommend the screening of autonomic dysfunction and antineuronal autoantibodies, as a test for patients with severe GI dysmotility. If criteria 1 and/or 2, 3 and/or 4 and/or 5 are present in the case, the case will be considered to have a high probability of AGID. Eventually, it is better to confirm the effect of immunotherapy on the case.
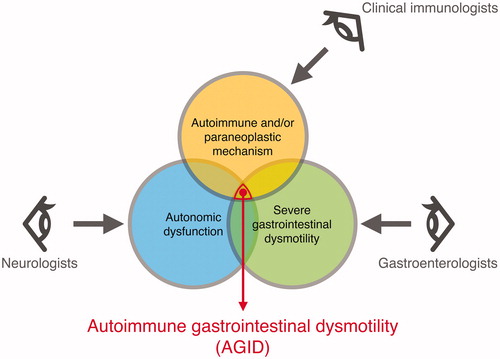
Neurogastroenterology, a subspecialty of gastroenterology that overlaps with neurology, was originally defined as the neurology of the digestive system and is related to the control of digestion via the ENS, CNS, and autonomic nervous system [Citation10]. Recent advancements in AGID research show that the conventional concept of neurogastroenterology is not sufficient for defining AGID, as AGID is now known to encompass both neurogastroenterology and clinical immunology (). Autoimmune neurology, which focuses on antibody-mediated autoimmune diseases including autoimmune encephalitis, autoimmune epilepsy, and autoimmune psychosis, is one of the most rapidly evolving fields in modern neurology [Citation93–96]. AGID should also be investigated as extensively as the abovementioned diseases, which will require cooperation among specialists, especially clinical immunologists, gastroenterologists, and neurologists.
Figure 5. Prospects of AGID. Collaboration among physicians and researchers should be encouraged to accelerate translational research, which can be applied to address the clinical and other challenges regarding AGID.
Acknowledgments
The authors are grateful to Drs. Hajime Isomoto, Hitomi Minami, Akio Ido, and Kazuhiko Nakao for useful discussions. We also thank Drs. Osamu Higuchi, Atsushi Kawakami, and Hidenori Matsuo for collaboration in the early stages of this work. The authors are indebted to members of the Kumamoto University Hospital Department of Neurology and Nagasaki Kawatana Medical Center Department of Neurology for discussions regarding this work.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Dhamija R, Tan KM, Pittock SJ, et al. Serologic profiles aiding the diagnosis of autoimmune gastrointestinal dysmotility. Clin Gastroenterol Hepatol. 2008;6(9):988–992.
- Lennon VA, Sas DF, Busk MF, et al. Enteric neuronal autoantibodies in pseudoobstruction with small-cell lung carcinoma. Gastroenterology. 1991;100(1):137–142.
- Sheikh SH, Shaw-Stiffel TA. The gastrointestinal manifestations of Sjögren's syndrome. Am J Gastroenterol. 1995;90:9–14.
- Flanagan EP, Saito YA, Lennon VA, et al. Immunotherapy trial as diagnostic test in evaluating patients with presumed autoimmune gastrointestinal dysmotility. Neurogastroenterol Motil. 2014;26(9):1285–1297.
- Mukaino A, Minami H, Isomoto H, et al. Anti-ganglionic AChR antibodies in Japanese patients with motility disorders. J Gastroenterol. 2018;53(12):1227–1240.
- Vernino S, Low PA, Fealey RD, et al. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med. 2000;343(12):847–855.
- Vernino S, Sandroni P, Singer W, et al. Invited article: autonomic ganglia: target and novel therapeutic tool. Neurology. 2008;70(20):1926–1932.
- Nakane S, Higuchi O, Koga M, et al. Clinical features of autoimmune autonomic ganglionopathy and the detection of subunit-specific autoantibodies to the ganglionic acetylcholine receptor in Japanese patients. PLoS One. 2015;10(3):e0118312.
- Fujikawa Y, Tominaga K. Neuro-gastroenterology: enteric nervous system. In: Tominaga K, editor. Functional dyspepsia: Evidences in Pathophysiology and Treatment. Singapore: Springer Nature; 2018. p. 51–58.
- Furness JB. The enteric nervous system and neurogastroenterology. Nat Rev Gastroenterol Hepatol. 2012;9(5):286–294.
- Benarroch EE. Enteric nervous system: functional organization and neurologic implications. Neurology. 2007;69(20):1953–1957.
- Rao M, Gershon MD. The bowel and beyond: the enteric nervous system in neurological disorders. Nat Rev Gastroenterol Hepatol. 2016;13(9):517–528.
- Vanner S, Greenwood-Van Meerveld B, Mawe G, et al. Fundamentals of neurogastroenterology: basic science. Gastroenterology. 2016;150(6):1280–1291.
- Hubball AW, Lang B, Souza MA, et al. Voltage-gated potassium channel (K(v) 1) autoantibodies in patients with chagasic gut dysmotility and distribution of K(v) 1 channels in human enteric neuromusculature (autoantibodies in GI dysmotility). Neurogastroenterol Motil. 2012;24(8):719–728.
- Hirano I, Pandolfino J. Chronic intestinal pseudo-obstruction. Dig Dis. 2000;18(2):83–92.
- Srinivasan S, Wiley JW. New insights into neural injury, repair, and adaptation in visceral afferents and the enteric nervous system. Curr Opin Gastroenterol. 2000;16:78–82.
- Goin JC, Sterin-Borda L, Bilder CR, et al. Functional implications of circulating muscarinic cholinergic receptor autoantibodies in chagasic patients with achalasia. Gastroenterology. 1999;117(4):798–805.
- De Giorgio R, Guerrini S, Barbara G, et al. Inflammatory neuropathies of the enteric nervous system. Gastroenterology. 2004;126(7):1872–1883.
- Jackson MW, Gordon TP, Waterman SA. Disruption of intestinal motility by a calcium channel-stimulating autoantibody in type 1 diabetes. Gastroenterology. 2004;126(3):819–828.
- Verne GN, Sallustio JE, Eaker EY. Anti-myenteric neuronal antibodies in patients with achalasia. A prospective study. Dig Dis Sci. 1997;42(2):307–313.
- Moses PL, Ellis LM, Anees MR, et al. Antineuronal antibodies in idiopathic achalasia and gastro-oesophageal reflux disease. Gut. 2003;52(5):629–636.
- Kraichely RE, Farrugia G, Pittock SJ, et al. Neural autoantibody profile of primary achalasia. Dig Dis Sci. 2010;55(2):307–311.
- Booy JD, Takata J, Tomlinson G, et al. The prevalence of autoimmune disease in patients with esophageal achalasia. Dis Esophagus. 2012;25(3):209–213.
- Quidute AR, Freitas EV, Lima TG, et al. Achalasia and thyroid disease: possible autoimmune connection? Arq Bras Endocrinol Metabol. 2012;56(9):677–682.
- Gyawall CP. Achalasia: new perspectives on an old disease. Neurogastroenterol Motil. 2016;28:4–11.
- Nojima Y, Mimura T, Hamasaki K, et al. Chronic intestinal pseudoobstruction associated with autoantibodies against proliferating cell nuclear antigen. Arthritis Rheum. 1996;39(5):877–879.
- Mok MY, Wong RW, Lau CS. Intestinal pseudo-obstruction in systemic lupus erythematosus: an uncommon but important clinical manifestation. Lupus. 2000;9(1):11–18.
- Viallard JF, Vincent A, Moreau JF, et al. Thymoma-associated neuromyotonia with antibodies against voltage-gated potassium channels presenting as chronic intestinal pseudo-obstruction. Eur Neurol. 2005;53(2):60–63.
- Nunokawa T, Yokogawa N, Ohtsuka H, et al. Transgastric long tube placement following percutaneous endoscopic gastrostomy for severe chronic intestinal pseudo-obstruction related to systemic sclerosis. Mod Rheumatol. 2015;25(6):958–961.
- Knowles CH, Lang B, Clover L, et al. A role for autoantibodies in some cases of acquired non-paraneoplastic gut dysmotility. Scand J Gastroenterol. 2002;37(2):166–170.
- Pasha SF, Lunsford TN, Lennon VA. Autoimmune gastrointestinal dysmotility treated successfully with pyridostigmine. Gastroenterology. 2006;131(5):1592–1596.
- Morimoto N, Takahashi S, Inaba T, et al. A case of seropositive autoimmune autonomic ganglionopathy with diffuse esophageal spasm. J Clin Neurosci. 2017;39:90–92.
- Kawanishi K, Moribata K, Kato J, et al. A case report of chronic intestinal pseudo-obstruction with autoimmune autonomic ganglionopathy suspected from seropositive results for anti-ganglionic acetylcholine receptor antibody. Nihon Shokakibyo Gakkai Zasshi. 2015;112(1):62–69.
- Yoshida T, Kinjo M, Nakane S. Autoimmune autonomic ganglionopathy associated with Sjögren's syndrome presenting with recurrent abdominal distension. BMJ Case Rep. 2018;2018:bcr2017223785.
- Nakane S, Mukaino A, Higuchi O, et al. Autoimmune autonomic ganglionopathy: an update on diagnosis and treatment. Expert Rev Neurother. 2018;18(12):953–965.
- Nakane S, Mukaino A, Higuchi O, et al. A comprehensive analysis of the clinical characteristics and laboratory features in 179 patients with autoimmune autonomic ganglionopathy. J Autoimmun. 2020;108:102403.
- Park K, Haberberger RV, Gordon TP, et al. Antibodies interfering with the type 3 muscarinic receptor pathway inhibit gastrointestinal motility and cholinergic neurotransmission in Sjögren's syndrome. Arthritis Rheum. 2011;63(5):1426–1434.
- McMahan ZH, Hummers LK. Gastrointestinal involvement in systemic sclerosis: diagnosis and management. Curr Opin Rheumatol. 2018;30(6):533–540.
- Emmanuel A. Current management of the gastrointestinal complications of systemic sclerosis. Nat Rev Gastroenterol Hepatol. 2016;13(8):461–472.
- Kumar S, Singh J, Rattan S, et al. Review article: pathogenesis and clinical manifestations of gastrointestinal involvement in systemic sclerosis. Aliment Pharmacol Ther. 2017;45(7):883–898.
- Ebert EC, Hagspiel KD. Gastrointestinal and hepatic manifestations of rheumatoid arthritis. Dig Dis Sci. 2011;56(2):295–302.
- Perlemuter G, Chaussade S, Wechsler B, et al. Chronic intestinal pseudo-obstruction in systemic lupus erythematosus. Gut. 1998;43(1):117–122.
- Nakane S, Umeda M, Kawashiri SY, et al. Detecting gastrointestinal manifestations in patients with systemic sclerosis using anti-gAChR antibodies. Arthritis Res Ther. 2020;22(32):1–10. DOI:10.1186/s13075-020-2128-z
- Howe CS, Eaker EY, Sallustio JE, et al. Antimyenteric neuronal antibodies in scleroderma. J Clin Invest. 1994;94(2):761–770.
- Eaker EY, Kuldau JG, Verne GN, et al. Myenteric neuronal antibodies in scleroderma: passive transfer evokes alterations in intestinal myoelectric activity in a rat model. J Lab Clin Med. 1999;133(6):551–556.
- Goldblatt F, Gordon TP, Waterman SA. Antibody-mediated gastrointestinal dysmotility in scleroderma. Gastroenterology. 2002;123(4):1144–1150.
- Nishigami E, Tochimoto A, Kawaguchi Y, et al. Characteristics of patients with early systemic sclerosis and severe gastrointestinal tract involvement. J Rheumatol. 2007;34:2050–2055.
- Kawaguchi Y, Nakamura Y, Matsumoto I, et al. Muscarinic-3 acetylcholine receptor autoantibody in patients with systemic sclerosis: contribution to severe gastrointestinal tract dysmotility. Ann Rheum Dis. 2009;68(5):710–714.
- Singh J, Mehendiratta V, Del Galdo F, et al. Immunoglobulins from scleroderma patients inhibit the muscarinic receptor activation in internal anal sphincter smooth muscle cells. Am J Physiol Gastrointest Liver Physiol. 2009;297(6):G1206–G1213.
- Kumar S, Singh J, Kedika R, et al. Role of muscarinic-3 receptor antibody in systemic sclerosis: correlation with disease duration and effects of IVIG. Am J Physiol Gastrointest Liver Physiol. 2016;310(11):G1052–G1060.
- Berger M, Steen VD. Role of anti-receptor autoantibodies in pathophysiology of scleroderma. Autoimmun Rev. 2017;16(10):1029–1035.
- McMahan ZH, Domsic RT, Zhu L, et al. Anti-RNPC-3 (U11/U12) antibodies in systemic sclerosis in patients with moderate-to-severe gastrointestinal dysmotility. Arthritis Care Res. 2019;71(9):1164–1170.
- Dunlop SP, Jenkins D, Spiller RC. Distinctive clinical, psychological, and histological features of postinfective irritable bowel syndrome. Am J Gastroenterol. 2003;98(7):1578–1583.
- Spiller RC. Postinfectious irritable bowel syndrome. Gastroenterology. 2003;124(6):1662–1671.
- Meeusen JW, Haselkorn KE, Fryer JP, et al. Gastrointestinal hypomotility with loss of enteric nicotinic acetylcholine receptors: active immunization model in mice. Neurogastroenterol Motil. 2013;25(1):84–88.
- Lennon VA, Ermilov LG, Szurszewski JH, et al. Immunization with neuronal nicotinic acetylcholine receptor induces neurological autoimmune disease. J Clin Invest. 2003;111(6):907–913.
- Vernino S, Low PA, Lennon VA. Experimental autoimmune autonomic neuropathy. J Neurophysiol. 2003;90(3):2053–2059.
- Vernino S, Ermilov LG, Sha L, et al. Passive transfer of autoimmune autonomic neuropathy to mice. J Neurosci. 2004;24(32):7037–7042.
- Sanchez-Ruiz M, Brunn A, Montesinos-Rongen M, et al. Enteric murine ganglionitis induced by autoimmune CD8 T cells mimics human gastrointestinal dysmotility. Am J Pathol. 2019;189(3):540–551.
- Drachman DB. Autonomic “myasthenia”: the case for an autoimmune pathogenesis. J Clin Invest. 2003;111(6):797–799.
- Spear ET, Holt EA, Joyce EJ, et al. Altered gastrointestinal motility involving autoantibodies in the experimental autoimmune encephalomyelitis model of multiple sclerosis. Neurogastroenterol Motil. 2018;30(9):e13349.
- Wunsch M, Jabari S, Voussen B, et al. The enteric nervous system is a potential autoimmune target in multiple sclerosis. Acta Neuropathol. 2017;134(2):281–295.
- Holtmann GJ, Ford AC, Talley NJ. Pathophysiology of irritable bowel syndrome. Lancet Gastroenterol Hepatol. 2016;1(2):133–146.
- Vich Vila A, Imhann F, Collij V, et al. Gut microbiota composition and functional changes in inflammatory bowel disease and irritable bowel syndrome. Sci Transl Med. 2018;10(472):eaap8914.
- Scheperjans F, Aho V, Pereira PA, et al. Gut microbiota are related to Parkinson's disease and clinical phenotype. Mov Disord. 2015;30(3):350–358.
- Cirstea MS, Yu AC, Golz E, et al. Microbiota composition and metabolism are associated with gut function in Parkinson's disease. Mov Disord. 2020. DOI:10.1002/mds.28052
- Mangiola F, Ianiro G, Franceschi F, et al. Gut microbiota in autism and mood disorders. World J Gastroenterol. 2016;22(1):361–368.
- Zhu X, Han Y, Du J, et al. Microbiota-gut-brain axis and the central nervous system. Oncotarget. 2017;8(32):53829–53838.
- Andréasson K, Alrawi Z, Persson A, et al. Intestinal dysbiosis is common in systemic sclerosis and associated with gastrointestinal and extraintestinal features of disease. Arthritis Res Ther. 2016;18(1):278.
- Godinho-Silva C, Filipa Cardoso F, Veiga-Fernandes H. Neuro-immune cell units: a new paradigm in physiology. Annu Rev Immunol. 2019;37:19–46.
- Tracey KJ. Reflex control of immunity. Nat Rev Immunol. 2009;9(6):418–428.
- Pavlov VA, Chavan SS, Tracey KJ. Molecular and functional neuroscience in immunity. Annu Rev Immunol. 2018;36:783–812.
- Teratani T, Mikami Y, Nakamoto N, et al. The liver-brain-gut neural arc maintains the Treg cell niche in the gut. Nature. 2020. DOI:10.1038/s41586-020-2425-3
- Chu C, Artis D, Chiu IM. Neuro-immune interactions in the tissues. Immunity. 2020;52(3):464–474.
- Ford AC, Talley NJ, Walker MM, et al. Increased prevalence of autoimmune diseases in functional gastrointestinal disorders: case-control study of 23471 primary care patients. Aliment Pharmacol Ther. 2014;40(7):827–834.
- Jones MP, Walker MM, Ford AC, et al. The overlap of atopy and functional gastrointestinal disorders among 23,471 patients in primary care. Aliment Pharmacol Ther. 2014;40(4):382–391.
- Koloski N, Jones M, Walker MM, et al. Population based study: atopy and autoimmune diseases are associated with functional dyspepsia and irritable bowel syndrome, independent of psychological distress. Aliment Pharmacol Ther. 2019;49(5):546–555.
- McMillan HJ, Srinivasan J. Achalasia, chronic sensory neuropathy, and N-type calcium channel autoantibodies: beneficial response to IVIG. Clin J Gastroenterol. 2010;3(2):78–82.
- Weinkauf C, McPhillips S, Krouse R, et al. Autoimmune gastrointestinal paralysis: failure of conventional treatment without immunomodulation. Case Rep Surg. 2014;2014:180654.
- Maier A, Mannartz V, Wasmuth H, et al. GAD antibodies as key link between chronic intestinal pseudoobstruction, autonomic neuropathy, and limb stiffness in a nondiabetic patient: a CARE-compliant case report and review of the literature. Medicine (Baltimore)). 2015;94(31):e1265.
- Greenburg DL, Mo CC, Hemmer PA. IVIG for thymoma-associated pseudo-obstruction: report of successful treatment. Eur Neurol. 2007;58(2):116–117.
- Gibbons CH, Vernino SA, Freeman R. Combined immunomodulatory therapy in autoimmune autonomic ganglionopathy. Arch Neurol. 2008;65(2):213–217.
- Iodice V, Kimpinski K, Vernino S, et al. Efficacy of immunotherapy in seropositive and seronegative putative autoimmune autonomic ganglionopathy. Neurology. 2009;72(23):2002–2008.
- Nishihara E, Koga M, Higuchi O, et al. Combined immunomodulatory therapies resulted in stepwise recovery in autoimmune autonomic ganglionopathy. Clin Exp Neuroimmunol. 2015;6(2):191–194.
- Sørhaug S, Steinshamn SL, Waldum HL. Octreotide treatment for paraneoplastic intestinal pseudo-obstruction complicating SCLC. Lung Cancer. 2005;48(1):137–140.
- Oton E, Moreira V, Redondo C, et al. Chronic intestinal pseudo-obstruction due to lymphocytic leiomyositis: is there a place for immunomodulatory therapy? Gut. 2005;54(9):1343–1344.
- Sodhi N, Camilleri M, Camoriano JK, et al. Autonomic function and motility in intestinal pseudoobstruction caused by paraneoplastic syndrome. Dig Dis Sci. 1989;34(12):1937–1942.
- Coret F, Bosca I, Fratalia L, et al. Long-lasting remission after rituximab treatment in a case of anti-Hu-associated sensory neuronopathy and gastric pseudoobstruction. J Neurooncol. 2009;93(3):421–423.
- Lautenbach E, Lichtenstein GR. Retroperitoneal leiomyosarcoma and gastroparesis: a new association and review of tumor-associated intestinal pseudo-obstruction. Am J Gastroenterol. 1341;90:1338.
- Lipowska AM, Micic D, Cavallo A, et al. Autoimmune gastrointestinal dysmotility due to small cell lung cancer. BMJ Case Rep. 2017;2017:bcr2017220890.
- Spear ET, Mawe GM. Enteric neuroplasticity and dysmotility in inflammatory disease: key players and possible therapeutic targets. Am J Physiol Gastrointest Liver Physiol. 2019;317(6):G853–G861.
- Bernardi MP, Warrier S, Lynch AC, et al. Acute and chronic pseudo-obstruction: a current update. ANZ J Surg. 2015;85(10):709–714.
- López-Chiriboga AS, Clardy SL. Emerging subspecialties in neurology: autoimmune neurology. Neurology. 2017;89(11):e129–e133.
- Graus F, Titulaer MJ, Balu R, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15(4):391–404.
- Sakamoto M, Matsumoto R, Togawa J, et al. Proposal of a diagnostic algorithm for autoimmune epilepsy: preliminary investigation of its utility. Rinsho Shinkeigaku. 2018;58(10):609–616.
- Pollak TA, Lennox BR, Müller S, et al. Autoimmune psychosis: an international consensus on an approach to the diagnosis and management of psychosis of suspected autoimmune origin. Lancet Psychiatry. 2020;7(1):93–108.