
Abstract
Rheumatoid arthritis (RA) is a systemic autoimmune mediated inflammatory disease characterized by progressive joint damage and extra-articular organ manifestations. Among the effector pathways and cells involved in the development of RA, activated B cells play a pivotal role in the pathological process of RA. P-glycoprotein (P-gp), a member of ATP-binding cassette transporters, is induced on the cell membrane by certain stimuli. P-gp transports various drugs from the cytoplasm to the cell exterior, resulting in the development of drug resistance. P-gp expression on B cells appears in patients with RA as the disease activity increases, and treatment of these patients’ results in modification of over-expression of P-gp on activated B cells. Evidence suggests that P-gp expressing-activated B cells play important roles in the pathogenesis and treatment resistance in RA through the efflux of intracellular drugs and progression of infiltration in inflammatory lesions. Therapies designed to target activated B cells might overcome refractory RA. Identification of the subsets of peripheral activated B cells that express P-gp in RA patients might help the selection of suitable treatment strategy.
1. Introduction
Rheumatoid arthritis (RA) is a systemic autoimmune-mediated inflammatory disease characterized by progressive destructive polyarthropathy with occasional systemic organ involvement [Citation1]. The treatment strategy centers on the control of immune abnormalities with disease modifying anti-rheumatic drugs (DMARDs) as soon as possible, to prevent progressive joint damage and extra-organ manifestations [Citation2]. However, some patients do not respond adequately to such treatments.
Among various effector pathways and cells, activated B cells play pivotal role in the pathogenesis of RA. In RA patients, these cells produce autoantibodies and various inflammatory cytokines, infiltrate the synovial tissues and present antigens to T cells [Citation3–5]. We reported previously that overexpression of P-glycoprotein (P-gp) on the pathologically activated B cells could lead to drug resistance and failure to control disease activity, especially in RA patients with high disease activity [Citation6–9]. In patients with refractory inflammatory bowel diseases (IBD), other investigators also reported upregulation of P-gp on peripheral lymphocytes [Citation10] and accumulation of highly activated plasmablasts in the peripheral blood [Citation11].
P-gp is a member of the ATP binding-cassette-transporters with two ATP binding sites, and functions as an energy-dependent transmembrane efflux pump. P-gp transports multiple drugs of P-gp substrates from the cytoplasm to the cell exterior through a process involving ATP hydrolysis, thus reducing their intracellular concentrations () [Citation12–19]. In other words, overexpression of P-gp on activated B cells can lead to the development of multidrug resistance.
Table 1. Relation of P-glycoprotein with disease modifying antirheumatic drugs and immunosuppressants.
Apart from its role in drug resistance, experimental and clinical evidence suggests the involvement of P-gp in the migration of various cancer cells and inflammatory cells. Overexpression of P-gp is associated with enhanced invasiveness and poor prognosis, whereas P-gp-specific inhibitors reduce migration of breast cancer cells and leukemia cells [Citation20–22]. The synovitis and RA-associated interstitial pneumonitis of patients with highly active RA show massive infiltration of C-X-C chemokine receptor 4 (CXCR4)-expressing P-gp+CD19+ B cells, together with the presence of CXCL12-expressing neovascular endothelial cells and fibroblasts [Citation9]. Furthermore, expansion of CXCR4+P-gp+CD19+ B cells in the peripheral blood reflect serious organ involvement [Citation9].
In this review, we discuss the relevance of P-gp-expressing activated B cells in treatment resistance and enhancement of tissue damage, and propose potentially effective treatments that target P-gp-expressing activated B cells for refractory RA.
2. Regulation of P-gp expression associated with enhancement of migration of B cells in RA
P-gp, also known as the ABC transporter subfamily B member 1 (ABCB1), is a 170-kDa product of the multidrug resistance-1 (MDR-1). P-gp is expressed congenitally on various endothelial cells, including the blood brain barrier, and on epithelial cells of various organs, including the gut, liver, kidney and pancreas and functions to protect cells from harmful toxic substances [Citation8]. Whereas resting normal lymphocytes show only marginal expression of P-gp, overexpression of this glycoprotein can be induced upon activation of lymphocytes by various stimuli [Citation8,Citation23]. For example, P-gp expression on activated B cells is directly regulated by activation of the Y-box binding protein-1 (YB-1), a MDR-1 transcription factor. Translocation of the YB-1 results in transcription of MDR-1 gene, which is induced by activation of the ERK pathway [Citation24–26] in response to immune stimuli, such as IL-6 [Citation23] and TNF-α [Citation8] which are associated with increased disease activity and play important pathogenic roles in inflammatory erosive arthritis in RA () [Citation5,Citation27–29]. The expression level of P-gp on B cells correlate with disease activity in patients with RA [Citation6]. Furthermore, treatment with corticosteroids without methotrexate (MTX) enhances P-gp expression on CD19+ B cells in RA patients with high disease activity [Citation6]. Thus, overexpression of P-gp on activated B cells is associated with poor control of disease activity in RA patients with highly active disease [Citation6–9].
Figure 1. Schematic diagram of induction of P-gp expression on B cells and P-gp+ B-cell targeting therapies in RA. (A) YB-1, which is located in the cytoplasm of resting B cells, is translocated by various signal transductions associated with activation of B cells, which results in overexpression of P-gp. (B) P-gp-related treatment resistance in RA can be probably overcome with P-gp+ B-cell targeting therapies as follows; inhibitors of B cell activation and differentiation (JAK inhibitors, TNF antagonists, IL-6 blockade, Anti-CD20 Monoclonal Antibodies), P-gp induction inhibitors (Methotrexate, TNF antagonists, IL-6 blockade, CXCR4 Antagonist), P-gp function inhibitors (P-glycoprotein Competitive Inhibitors) and P-gp+ B-cells depletors (Anti-CD20 Monoclonal Antibodies).
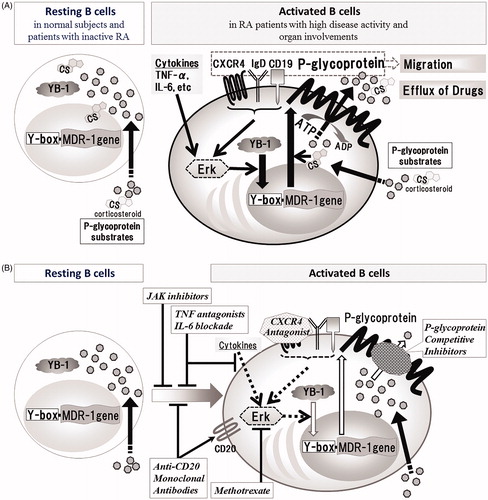
The P-gp+ B cell subset that co-expresses CXCR4 are closely related to RA disease activity and serious organ involvement [Citation9]. The upstream CXCR4 gene contains a putative consensus Y-box-binding site (inverted CCAAT box) to which the MDR-1 transcription factor YB-1 can bind [Citation30]. It remains unclear whether activation of YB-1 is directly involved in the upregulation of CXCR4 gene. However, a Syk-dependent IgD-BCR signal, which is triggered by actin cytoskeleton remodeling following CXCL12/CXCR4 axis activation, is initiated and results in induction of Ca+ influx and activation of the ERK pathway [Citation31]. This could lead to induction of P-gp expression through ERK pathway. Fragmented hyaluronan, one of inflammatory extracellular matrix, induces P-glycoprotein expression on lymphocytes through CD44 [Citation32]. Colone et al. [Citation20] reported that P-gp interacts with CD44 through the activation of the ERK and MAPK pathways and results in increase of the invasive behavior, associated with an increase in MMP production and proteolytic activity. CD44 did not increase the invasive behavior in the absence of P-gp. Thus, P-gp may cooperate with molecules involved in induction of own expression and result in increase of migration and invasive potential. P-gp+CXCR4+CD19+ B cells seems to have high-migration capacity and can enhance pathological lesions. In addition to B cells, plasmablasts also coexpress CXCR4 and CD19 [Citation33]. Evidence suggests the involvement of CXCL12/CXCR4 axis in the migration of plasmablasts to the inflamed tissues [Citation34] and the association of P-gp+CXCR4+CD19+ B cells with severe organ injury in RA patients [Citation9,Citation35]. The expression and binding capacity of CXCL12 are increased in the RA synovium in the presence of TNF-α, a cytokine known to induce P-gp [Citation36,Citation37]. In addition to RA, the CXCL12/CXCR4 axis also plays a role in the migration of inflammatory cells in other autoimmune diseases [Citation35,Citation38,Citation39]. For example, marked accumulation of CXCR4+CD19+ B cells was reported in the inflamed mucosa of ulcerative colitis (UC) [Citation38] and significantly high CXCR4 and CXCL12 mRNA levels were reported in bronchoalveolar lavages of pulmonary sarcoidosis [Citation39].
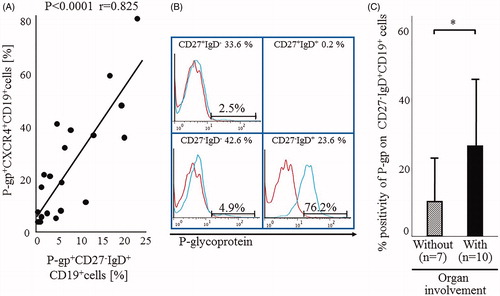
CXCR4 is located in proximity to the IgD-B-cell antigen receptor (BCR), which colocalizes with the coreceptor CD19, and IgD-BCR, CD19 and CXCR4 are functionally linked during the induction of B-cell migration associated with CXCL12-mediated CXCR4 signaling [Citation31]. The lipid rafts on the plasma membrane contain constitutively associated CD19 and P-gp molecules [Citation40]. We have reported previously the presence of strong and significant correlation between the proportions of P-gp+CD27-IgD+CD19+ B cells and P-gp+CXCR4+CD19+ B cells in RA patients (). shows images from a representative patient with highly active RA and extra-articular involvement who showed expansion of P-gp+CXCR4+CD19+ B cells and preferentially high P-gp expression on CD27-IgD+CD19+ B cells. Therefore, majority of P-gp+CXCR4+CD19+ B cells might contain P-gp+CD27-IgD+CD19+ B cells. Thus, P-gp+CD27-IgD+CD19+ B cells were also significantly increased in RA patients with extra-articular involvements (). Other studies also showed significant upregulation of P-gp expression on peripheral lymphocytes and accumulation of antigen-selected IgD-only B cells in the bronchial mucosa of asthmatic patients with high serum IgD levels [Citation41–43]. CD19+ B cells expressing triplets of P-gp-CXCR4-IgD may enhance the migration activity followed by progression of infiltration in CXCL12-expressing inflammatory lesions.
Figure 2. Relations among P-gp+CXCR4+ B cells, P-gp+ CD27-IgD+B cells and RA organ involvement. (A) Correlation between the proportions of P-gp+CXCR4+ B cells and P-gp+ CD27-IgD+B cells in RA patients. Statistical analysis was performed by Person’s correlation analysis. (B) Flow cytometric analysis identified P-gp+ CD27-IgD+B cells in the representative RA patient with rheumatoid vasculitis. Values at the top of each section are percentages of CD19+B cell subpopulations based on CD27/IgD classification. Flow cytometric analysis showed P-gp expression on each B cell subpopulation (blue lines). Data represent the percentages of P-gp-positively stained B cell subpopulations. Red: isotype-control FITC-conjugated anti-mouse IgG Ab. The number of CD27+IgD+CD19+ B cells was lower than that was available for the histogram exhibition and P-gp expression analysis. (C) Flow cytometry for P-gp+ CD27-IgD+B cells in 17 RA patients, including 10 with (closed bar) and 7 without (hatched bar) organ involvement. Values are mean ± SD of independent experiments. *p < .05, by non-paired t-test. RA disease activity, as estimated by the SDAI score, was not significantly different between the two groups (with: 26.5% ± 8.3, without: 30.9% ± 12.9; p = .41). Organ involvement included interstitial pneumonia (n = 2), interstitial pneumonia with rheumatoid vasculitis (n = 3), Felty syndrome (n = 1), amyloidosis (n = 1) and lymphadenopathy (n = 1). (A–C) Specific antibodies used for staining and flow cytometric analysis, including MRK16 for P-gp (a specific mAb against P-gp; Kyowa Medex, Tokyo) with FITC-conjugated goat anti-mouse IgG mAb, cy-chrome-conjugated CD19 mAb, APC-conjugated CD27 mAb and PE-conjugated IgD and CXCR4 mAb (BD Biosciences Pharmingen).
In patients with RA, B cells start to express P-gp with the increase in disease activity, and the expression level on activated B cells is further enhanced in RA patients with high disease activity treated with corticosteroids but without MTX. Furthermore, the P-gp overexpression on activated CD19+ B cells associated with CXCR4/IgD expression subsequently results in treatment resistance and progressive destructive arthritis with extra-articular involvements.
3. Potential of treatments targeting P-gp+ B cell compartments in RA
As reviewed above, P-gp+CD19+B cells, especially those co-expressing CXCR4 and IgD, are pro-inflammatory activated B cells associated with drug resistance, disease activity and progression of inflammatory lesions in RA. Therefore, P-gp+ B cells targeting therapies may be beneficial in the control of disease activity in refractory RA ().
3.1. Conventional synthetic DMARDs
MTX is one of the anchor drugs used for the treatment of patients with RA. It acts by inhibiting the proliferation and differentiation of B cells and reducing the production of immunoglobulins, including rheumatoid factor [Citation44]. MTX is known to reduce the activation of ERK and production of cytokines, including IL-6 and TNF-α [Citation45,Citation46]. Thus, MTX seems to inhibit both B cells activation and the ERK pathway and that such inhibition may result in downregulation of P-gp expression on B cells. In fact, MTX is reported to limit P-gp expression on B cells in patients with highly active RA [Citation6], as well as overcome steroid resistance in sarcoidosis [Citation47].
Tacrolimus (TAC) acts as a calcineurin inhibitor and inhibits the differentiation of naive T cells into functional pd1+iCOS+Tfh-like cells, resulting in inhibition of Tfh-dependent B-cell proliferation and differentiation into plasma cells and transitional B cells [Citation48]. TAC also acts as a P-gp competitive inhibitor with the potential of overcoming drug-resistance, similar to cyclosporine A [Citation8,Citation49]. We have demonstrated that treatment with TAC resulted in recovery of intracellular concentrations of dexamethasone, a representative P-gp substrate, in IL-2-activated lymphocytes using doses lower than the trough level measured clinically when TAC is used as a calcineurin inhibitor (). In fact, the presence of low concentrations of TAC in cultures of lymphocytes harvested from highly active RA patients, induced recovery of intra-lymphocytes dexamethasone concentrations [Citation8]. The efficacy of TAC in the treatment of patients with RA depends on both the expression level of P-gp on lymphocytes and its inhibitory effect on the drug exclusion function of P-gp [Citation49]. Thus, P-gp competitors, including TAC regulate P-gp-mediated drug resistance by recovery of intracellular P-gp substrates (). Other investigators also reported the efficacy of TAC in asthma and interstitial pneumonia associated with autoimmune diseases [Citation50,Citation51]. Patients with myasthenia gravis exhibit increased P-gp function in lymphocytes, particularly in refractory patients compared with corticosteroid-responders, whereas it is attenuated by TAC treatment [Citation52,Citation53].
Figure 3. Tacrolimus inhibits excretion of intracellular dexamethasone through P-glycoprotein. 1 × 106 of PBMCs were pre-incubated with (solid circles) or without (open circle) 10 ng/mL of IL-2 for 4 h. Then, 20-min after the addition of [6,7-3H(N)]-dexamethasone and [14C] n-butanol, the cell to medium ratio (C/M ratio; an index of intracellular and extracellular dexamethasone concentration ratio) was evaluated in the presence of the indicated concentrations of tacroliums. The C/M ratio was computed using the following formula: C/M ratio = [(3H in cell fraction/14C in cell fraction)/(3H in medium fraction/14C in medium fraction)]. Data are mean ± SD of five independent experiments. *p < .05, **p < .01, by one-way ANOVA.
![Figure 3. Tacrolimus inhibits excretion of intracellular dexamethasone through P-glycoprotein. 1 × 106 of PBMCs were pre-incubated with (solid circles) or without (open circle) 10 ng/mL of IL-2 for 4 h. Then, 20-min after the addition of [6,7-3H(N)]-dexamethasone and [14C] n-butanol, the cell to medium ratio (C/M ratio; an index of intracellular and extracellular dexamethasone concentration ratio) was evaluated in the presence of the indicated concentrations of tacroliums. The C/M ratio was computed using the following formula: C/M ratio = [(3H in cell fraction/14C in cell fraction)/(3H in medium fraction/14C in medium fraction)]. Data are mean ± SD of five independent experiments. *p < .05, **p < .01, by one-way ANOVA.](/cms/asset/e98f710d-b580-4e3b-8336-0acbc60c5dbb/timm_a_1825276_f0003_b.jpg)
3.2. Targeted synthetic DMARDs
B-cells are activated by signal transduction through common γ-chain cytokine receptors. Janus kinase (JAK) inhibitors, which impair signal transduction through the common γ-chain, inhibit plasmablast development, production of IL-6 and the secretion of antibodies from B-cells [Citation54,Citation55]. The majority of JAK inhibitors are substrates of P-gp () [Citation18,Citation56]. In vitro studies demonstrated attenuation of the effects of JAK inhibitors in stimulated peripheral blood B cells [Citation54]. Whereas, first anchor drug MTX is known to inhibit P-gp expression on B cells [Citation6], thus, MTX might prolong the effects of combined JAK inhibitors by preventing P-gp-related attenuation of the effects. Actually, the combination of JAK inhibitors and MTX is especially effective in patients with refractory RA who otherwise respond poorly to biological DMARDs or conventional synthetic DMARDs [Citation57,Citation58]. Since B cell activation induced by IL-4 via JAK signaling aggravates experimental asthma, several clinical trials have highlighted recently the potential therapeutic benefits of inhaled JAK inhibitors in asthmatic patients [Citation59,Citation60].
3.3. Biological DMARDs
Biological DMARDs (bDMARDs) act extracellularly, and the molecular weights of bDMARDs greatly exceed the molecular weights of the substrates of P-gp which are ranged 300–2000 Da [Citation61]. Therefore, bDMARDs inhibit lymhocyte activation without being affected by P-gp [Citation8]. TNF antagonists and IL-6 blockers inhibit B cell trafficking towards inflammatory sites in RA [Citation62]. The former group of agents also reduces P-gp expression on B cells in RA patients [Citation6–9].
The use of TNF antagonists for the treatment of refractory RA with organ involvement is reported to result in elimination of P-gp+CXCR4+CD19+ B cells with subsequent control of the pathological process [Citation9]. On the other autoimmune diseases, the clinical response to TNF antagonists in patients with inflammatory bowel diseases (IBD) is reported to be obtained through the effects to B cells. The IL10-producing B cells of both ulcerative colitis and Crohn disease patients were reduced compared with those in healthy control patients and detected a significant increase in responding patients after treatment with infliximab, a TNF antagonist [Citation63]. IgD+CD27- B cell and activated B cell subsets in reported to be predominant in peripheral blood of sarcoidosis patients [Citation64]. Treatment of patients with CNS sarcoidosis unresponsive to immunosuppressants, using infliximab resulted in improvement in imaging and clinical findings [Citation65].
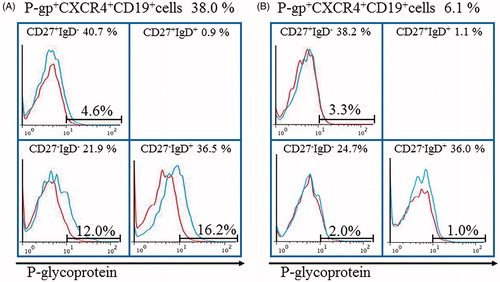
Other studies also reported the clinical benefits of tocilizumab, an IL-6 blocker, in RA patients, which induced/expanded B regulatory cells [Citation66], and in SLE by restoration of B and T cell homoeostasis, together with normalization of abnormal B and T cell subsets [Citation67]. We analyzed the changes in P-gp expressions on B cells in RA patient responding to tocilizumab therapy. The RA patient with microscopic polyangiitis was treated with high-dose corticosteroid and intravenous cyclophosphamide pulse therapy during a period of 3 months but showed exacerbation of RA disease activity to DAS28 of 5.3 by prednisolone (PSL) tapering to 25 mg/day. She showed accumulation of peripheral P-gp+CXCR4+ B cells and P-gp+IgD+ B cells (). The treatment was switched to tocilizumab, in addition to oral PSL, and resulted in significant fall in peripheral P-gp+CXCR4+ B cells and P-gp+IgD+ B cells within 4 weeks (), together with improvement of synovitis to DAS28 of 3.6 and tapering to 15 mg/day of PSL and without relapse of microscopic polyangiitis. Treatment of patients with systemic lupus erythematosus (SLE) using tocilizumab resulted in inhibition of B cell differentiation and suppression of peripheral plasma cell compartments [Citation68]. These findings were assumed to be mediated through downregulation of P-gp expression on activated B cells since tocilizumab blocks IL-6 as the P-gp inducer [Citation23]. Tocilizumab was also found to reduce plasmablasts in peripheral blood, AQP4-IgG titers and relapse activity in at least some patients with neuromyelitis optica (NMO) who showed activation of B cells accompanied by production of AQP4-reactive IgGs [Citation69].
Figure 4. Tocilizumab downregulates P-gp+CXCR4+B cells and P-gp+CD27-IgD+B cells in RA with microscopic polyangiitis. Flow cytometric analysis identified P-gp+CXCR4+B cells and P-gp+CD27-IgD+B cells in the RA patient. Values above the chart are the proportions of P-gp+CXCR4+ B cells just at start (A) and 4 weeks after (B) of tocilizumab therapy. Values at the top of each section are percentages of CD19+B cell subpopulations based on CD27/IgD classification. Flow cytometric analysis showed P-gp expression on each B cell subpopulation (blue lines). Data represent the percentages of P-gp-positively stained B cell subpopulations. Red: isotype-control FITC-conjugated anti-mouse IgG Ab. The number of CD27+IgD+CD19+ B cells was lower than that was available for the histogram exhibition and P-gp expression analysis. Specific antibodies used for staining and flow cytometric analysis, including MRK16 for P-gp (a specific mAb against P-gp; Kyowa Medex, Tokyo) with FITC-conjugated goat anti-mouse IgG mAb, cy-chrome-conjugated CD19 mAb, APC-conjugated CD27 mAb and PE-conjugated IgD and CXCR4 mAb (BD Biosciences Pharmingen).
Another B cell depletion therapy is the use of anti-CD20 monoclonal antibody (e.g. rituximab). This treatment is largely designed to deplete CD20+ B cells, including pre-B cells, immature B cells, IgD+ native B cells and memory B cells and results in impaired generation of CD20-plasmablasts and low titers of autoantibodies, such as rheumatoid factors and anti-citrullinated protein/peptide antibodies [Citation70,Citation71]. Adlowitz et al. [Citation72] reported that RA patients who received B cell depletion therapy showed marked reduction of B cells-TNF production and that such reduction correlated with reduction in memory B cells. B cell depletion and reconstitution may lead to improvement in immune complex-dependent synovitis by reduction of autoantibody production, and also long-term remission by reduced populations of pro-inflammatory activated B cells. Treatment with rituximab has been reported to be efficacious also in corticosteroids-resistant patients with pulmonary sarcoidosis, neurosarcoidosis and refractory NMO [Citation64,Citation73].
3.4. CXCR4 antagonists
CXCR4 antagonists significantly suppress delayed-type hypersensitivity induced by sheep red blood cells, and significantly reduce disease severity experimental collagen-induced arthritis in mice. The use of CXCR4 antagonists could be useful for RA-associated interstitial pneumonia. CXCR4 antagonists have been reported to significantly decrease dissemination of diffuse large B cell lymphoma (DLBCL) and suppress pulmonary metastasis from breast cancer and melanoma cells in mice [Citation35,Citation74]. CXCR4 antagonist synergistically enhanced the anti-proliferative/pro-apoptotic effect of rituximab on DLBCL cells and was suggested to improve treatment outcome for DLBCL patients [Citation75]. CXCR4 antagonist ameliorated colonic inflammation in murine experimental IBD including DSS-induced colitis and IL-10 KO, accompanied by reduction in TNF production from mesenteric lymph node cells [Citation76].
4. Conclusion
P-gp+CD19+ B cells that co-express CXCR4 and IgD are pro-inflammatory activated B cells that play a pathological role in the development of destructive arthritis with extra-articular involvement and the acquisition of P-gp-mediated multidrug resistance. Accordingly, measurement of the percentage of peripheral P-gp+CXCR4+IgD+CD19+ B cells may help in the recognition of drug resistance and disease severity, and help in the design of new treatment modality for RA patients with highly active disease.
Acknowledgement
The authors thank Ms. T. Adachi for the excellent technical assistance.
Disclosure statement
Y. Tanaka has received consulting fees, lecture fees and/or honoraria from Abbvie, Chugai, Daiichi-Sankyo, Bristol-Myers, Mitsubishi-Tanabe, Astellas, Takeda, Pfizer, Teijin, Asahi-kasei, YL Biologics, Sanofi, Janssen, Eli Lilly, GlaxoSmithKline and also research grants from Mitsubishi-Tanabe, Takeda, Daiichi-Sankyo, Chugai, Bristol-Myers, MSD, Astellas, Abbvie, Eisai. S. Tsujimura declares no conflict of interest.
Additional information
Funding
References
- Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet. 2010;376:1094–1108.
- Smolen JS, Landewé R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76(6):960–977.
- Moura RA, Graca L, Fonseca JE. To B or not to B the conductor of rheumatoid arthritis orchestra. Clin Rev Allergy Immunol. 2012;43(3):281–291.
- Duddy ME, Alter A, Bar-Or A. Distinct profiles of human B cell effector cytokines: a role in immune regulation? J Immunol. 2004;172(6):3422–3427.
- Gottenberg JE, Dayer JM, Lukas C, et al. Serum IL-6 and IL-21 are associated with markers of B cell activation and structural progression in early rheumatoid arthritis: results from the ESPOIR cohort. Ann Rheum Dis. 2012;71(7):1243–1248.
- Tsujimura S, Saito K, Nawata M, et al. Overcoming drug resistance induced by P-glycoprotein on lymphocytes in patients with refractory rheumatoid arthritis. Ann Rheum Dis. 2008;67(3):380–388.
- Tsujimura S, Saito K, Nakayamada S, et al. Etanercept overcomes P-glycoprotein-induced drug resistance in lymphocytes of patients with intractable rheumatoid arthritis. Mod Rheumatol. 2010;20(2):139–146.
- Tsujimura S, Tanaka Y. Disease control by regulation of P-glycoprotein on lymphocytes in patients with rheumatoid arthritis. World J Exp Med. 2015;5(4):225–231.
- Tsujimura S, Adachi T, Saito K, et al. Relevance of P-glycoprotein on CXCR4+ B cells to organ manifestation in highly active rheumatoid arthritis. Mod Rheumatol. 2018;28(2):276–286.
- Farrell RJ, Murphy A, Long A, et al. High multidrug resistance (P-glycoprotein 170) expression in inflammatory bowel disease patients who fail medical therapy. Gastroenterology. 2000;118(2):279–288.
- Noble A, Durant L, Hoyles L, et al. Deficient resident memory T-cell and Cd8 T-cell response to commensals in inflammatory bowel disease. J Crohns Colitis. 2019;26:1–13.
- Leonard GD, Polgar O, Bates SE. ABC transporters and inhibitors: new targets, new agents. Curr Opin Investig Drugs. 2002;3(11):1652–1659.
- McKeegan KS, Borges-Walmsley MI, Walmsley AR. The structure and function of drug pumps: an update. Trends Microbiol. 2003;11(1):21–29.
- Senarathna SM, Page-Sharp M, Crowe A. The interactions of P-glycoprotein with antimalarial drugs, including substrate affinity, inhibition and regulation. PLoS One. 2016;11(4):e0152677.
- Lin JH. Drug-drug interaction mediated by inhibition and induction of P-glycoprotein. Adv Drug Deliv Rev. 2003;55(1):53–81.
- Medeiros M, Castañeda-Hernández G, Ross CJ, et al. Use of pharmacogenomics in pediatric renal transplant recipients. Front Genet. 2015;6:41.
- Gwak EH, Yoo HY, Kim SH. Effects of diabetes mellitus on the disposition of tofacitinib, a janus kinase inhibitor, in rats. Biomol Ther (Seoul). 2020;28(4):361–369.
- Posada MM, Cannady EA, Payne CD, et al. Prediction of transporter-mediated drug-drug interactions for baricitinib. Clin Transl Sci. 2017;10(6):509–519.
- Zhu T, Howieson C, Wojtkowski T, et al. The effect of verapamil, a P-glycoprotein inhibitor, on the pharmacokinetics of peficitinib, an orally administered, once-daily JAK inhibitor. Clin Pharmacol Drug Dev. 2017;6(6):548–555.
- Colone M, Calcabrini A, Toccacieli L, et al. The multidrug transporter P-glycoprotein: a mediator of melanoma invasion? J Invest Dermatol. 2008;128(4):957–971.
- Zhang F, Zhang H, Wang Z, et al. P-glycoprotein associates with Anxa2 and promotes invasion in multidrug resistant breast cancer cells. Biochem Pharmacol. 2014;87(2):292–302.
- Hu M, Liu Y, Deng C, et al. Enhanced invasiveness in multidrug resistant leukemic cells is associated with overexpression of P-glycoprotein and cellular inhibitor of apoptosis protein. Leuk Lymphoma. 2011;52(7):1302–1311.
- List AF, Kopecky KJ, Willman CL, et al. Cyclosporine inhibition of P-glycoprotein in chronic myeloid leukemia blast phase. Blood. 2002;100(5):1910–1912.
- Tsujimura S, Kawabe A, Tanaka Y. P-glycoprotein expressing-B cell associated active true renal lupus vasculitis in lupus nephritis. Lupus: Open Access. 2020;5:1–6.
- Zhao BX, Sun YB, Wang SQ, et al. Grape seed procyanidin reversal of p-glycoprotein associated multi-drug resistance via down-regulation of NF-κB and MAPK/ERK mediated YB-1 activity in A2780/T cells. PLoS One. 2013;8(8):e71071.
- Tomiyasu H, Watanabe M, Sugita K, et al. Regulations of ABCB1 and ABCG2 expression through MAPK pathways in acute lymphoblastic leukemia cell lines. Anticancer Res. 2013;33(12):5317–5323.
- Shen H, Xu W, Luo W, et al. Upregulation of mdr1 gene is related to activation of the MAPK/ERK signal transduction pathway and YB-1 nuclear translocation in B-cell lymphoma. Exp Hematol. 2011;39(5):558–569.
- Feng W, Liu H, Luo T, et al. Combination of IL-6 and sIL-6R differentially regulate varying levels of RANKL-induced osteoclastogenesis through NF-κB, ERK and JNK signaling pathways. Sci Rep. 2017;7:41411.
- Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med. 2001;344(12):907–916.
- Basaki Y, Hosoi F, Oda Y, et al. Akt-dependent nuclear localization of Y-box-binding protein 1 in acquisition of malignant characteristics by human ovarian cancer cells. Oncogene. 2007;26(19):2736–2746.
- Becker M, Hobeika E, Jumaa H, et al. CXCR4 signaling and function require the expression of the IgD-class B-cell antigen receptor. Proc Natl Acad Sci USA. 2017;114(20):5231–5236.
- Tsujimura S, Saito K, Kohno K, et al. Fragmented hyaluronan induces transcriptional up-regulation of the multidrug resistance-1 gene in CD4+ T cells. J Biol Chem. 2006;281(49):38089–38097.
- Odendahl M, Mei H, Hoyer BF, et al. Generation of migratory antigen-specific plasma blasts and mobilization of resident plasma cells in a secondary immune response. Blood. 2005;105(4):1614–1621.
- Pak HK, Nam B, Lee YK, et al. Human plasmablast migration toward CXCL12 requires glucose oxidation by enhanced pyruvate dehydrogenase activity via AKT. Front Immunol. 2018;9:1742.
- Tamamura H, Tsutsumi H, Masuno H, et al. Development of low molecular weight CXCR4 antagonists by exploratory structural tuning of cyclic tetra- and pentapeptide-scaffolds towards the treatment of HIV infection, cancer metastasis and rheumatoid arthritis. Curr Med Chem. 2007;14(1):93–102.
- Nanki T, Nagasaka K, Hayashida K, et al. Chemokines regulate IL-6 and IL-8 production by fibroblast-like synoviocytes from patients with rheumatoid arthritis. J Immunol. 2001;167(9):5381–5385.
- Santiago B, Baleux F, Palao G, et al. CXCL12 is displayed by rheumatoid endothelial cells through its basic amino-terminal motif on heparan sulfate proteoglycans . Arthritis Res Ther. 2006;8(2):R43.
- Uo M, Hisamatsu T, Miyoshi J, et al. Mucosal CXCR4+ IgG plasma cells contribute to the pathogenesis of human ulcerative colitis through FcγR-mediated CD14 macrophage activation. Gut. 2013;62(12):1734–1744.
- Antoniou KM, Soufla G, Proklou A, et al. Different activity of the biological axis VEGF-Flt-1 (fms-like tyrosine kinase 1) and CXC chemokines between pulmonary sarcoidosis and idiopathic pulmonary fibrosis: a bronchoalveolar lavage study. Clin Dev Immunol. 2009;2009:537929.
- Ghetie MA, Marches R, Kufert S, et al. An anti-CD19 antibody inhibits the interaction between P-glycoprotein (P-gp) and CD19, causes P-gp to translocate out of lipid rafts, and chemosensitizes a multidrug-resistant (MDR) lymphoma cell line. Blood. 2004;104(1):178–183.
- Kopriva F, Dzubak P, Potesil J, et al. The anti-inflammatory effects of inhaled corticosteroids versus anti-leukotrienes on the lymphocyte P-glycoprotein (PGP) expression in asthmatic children. J Asthma. 2009;46(4):366–370.
- Salpietro DC, Masaracchio A, Turiaco A, et al. Serum IgD levels in children with atopic asthma. A longitudinal study. Minerva Pediatr. 2001;53:1–5.
- Ohm-Laursen L, Meng H, Chen J, et al. Local clonal diversification and dissemination of B lymphocytes in the human bronchial mucosa. Front Immunol. 1976;9:1976.
- Glaesener S, Quách TD, Onken N, et al. Distinct effects of methotrexate and etanercept on the B cell compartment in patients with juvenile idiopathic arthritis. Arthritis Rheumatol. 2014;66(9):2590–2600.
- Wessels JA, Huizinga TW, Guchelaar HJ. Recent insights in the pharmacological actions of methotrexate in the treatment of rheumatoid arthritis. Rheumatology (Oxford). 2008;47(3):249–255.
- Yang X, Zhao Y, Jia X, et al. CP-25 combined with MTX/ LEF ameliorates the progression of adjuvant-induced arthritis by the inhibition on GRK2 translocation. Biomed Pharmacother. 2019;110:834–843.
- Hilderson I, Van Laecke S, Wauters A, et al. Treatment of renal sarcoidosis: is there a guideline? Overview of the different treatment options. Nephrol Dial Transplant. 2014;29(10):1841–1847.
- Kraaijeveld R, Li Y, Yan L, et al. Inhibition of T helper cell differentiation by tacrolimus or sirolimus results in reduced B-cell activation: effects on t follicular helper cells. Transplant Proc. 2019;51(10):3463–3473.
- Suzuki K, Saito K, Tsujimura S, et al. Tacrolimus, a calcineurin inhibitor, overcomes treatment unresponsiveness mediated by P-glycoprotein on lymphocytes in refractory rheumatoid arthritis. J Rheumatol. 2010;37(3):512–520.
- Kandikattu HK, Mishra A. Immunomodulatory effects of tacrolimus (FK506) for the treatment of allergic diseases. Int J Cell Biol Physiol. 2018;1(1–2):5–13.
- Ando M, Miyazaki E, Yamasue M, et al. Successful treatment with tacrolimus of progressive interstitial pneumonia associated with amyopathic dermatomyositis refractory to cyclosporine. Clin Rheumatol. 2010;29(4):443–445.
- Richaud-Patin Y, Vega-Boada F, Vidaller A, et al. Multidrug resistance-1 (MDR-1) in autoimmune disorders IV. P-glycoprotein overfunction in lymphocytes from myasthenia gravis patients. Biomed Pharmacother. 2004;58(5):320–324.
- Tanaka S, Hirano T, Saito T, et al. P-glycoprotein function in peripheral blood mononuclear cells of myasthenia gravis patients treated with tacrolimus. Biol Pharm Bull. 2007;30(2):291–296.
- Rizzi M, Lorenzetti R, Fischer K, et al. Impact of tofacitinib treatment on human B-cells in vitro and in vivo. J Autoimmun. 2017;77:55–66.
- Kubo S, Nakayamada S, Sakata K, et al. Janus kinase inhibitor Baricitinib modulates human innate and adaptive immune system. Front Immunol. 2018;9:1510.
- Durmus S, Xu N, Sparidans RW, et al. P-glycoprotein (MDR1/ABCB1) and breast cancer resistance protein (BCRP/ABCG2) restrict brain accumulation of the JAK1/2 inhibitor, CYT387. Pharmacol Res. 2013;76:9–16.
- Fleischmann R, Mysler E, Hall S, et al. Efficacy and safety of tofacitinib monotherapy, tofacitinib with methotrexate, and adalimumab with methotrexate in patients with rheumatoid arthritis (ORAL Strategy): a phase 3b/4, double-blind, head-to-head, randomised controlled trial. Lancet. 2017;390(10093):457–468.
- Taylor PC, Lee YC, Fleischmann R, et al. Achieving pain control in rheumatoid arthritis with baricitinib or adalimumab plus methotrexate: results from the RA-BEAM Trial. J Clin Med. 2019;8(6):831.
- Xia F, Deng C, Jiang Y, et al. IL4 (interleukin 4) induces autophagy in B cells leading to exacerbated asthma. Autophagy. 2018;14(3):450–464.
- Zak M, Dengler HS, Rajapaksa NS. Inhaled Janus Kinase (JAK) inhibitors for the treatment of asthma. Bioorg Med Chem Lett. 2019;29(20):126658.
- Tsujimura S, Saito K, Nakayamada S, et al. Relevance of multidrug resistance 1 and P-glycoprotein to drug resistance in patients with systemic lupus erythematosus. Histol Histopathol. 2007;22(4):465–468.
- Moura RA, Quaresma C, Vieira AR, et al. B-cell phenotype and IgD-CD27- memory B cells are affected by TNF-inhibitors and tocilizumab treatment in rheumatoid arthritis. PLoS One. 2017;12(9):e0182927.
- Schnell A, Schwarz B, Wahlbuhl M, et al. Distribution and cytokine profile of peripheral B cell subsets is perturbed in pediatric IBD and partially restored during a successful IFX therapy. Inflamm Bowel Dis. 2020;izaa054.
- Kudryavtsev I, Serebriakova M, Starshinova A, et al. Imbalance in B cell and T follicular helper cell subsets in pulmonary sarcoidosis. Sci Rep. 2020;10(1):1059.
- Gelfand JM, Bradshaw MJ, Stern BJ, et al. Infliximab for the treatment of CNS sarcoidosis: a multi-institutional series. Neurology. 2017;89(20):2092–2100.
- Snir A, Kessel A, Haj T, et al. Anti-IL-6 receptor antibody (tocilizumab): a B cell targeting therapy. Clin Exp Rheumatol. 2011;29:697–700.
- Illei GG, Shirota Y, Yarboro CH, et al. Tocilizumab in systemic lupus erythematosus: data on safety, preliminary efficacy, and impact on circulating plasma cells from an open-label phase I dosage-escalation study. Arthritis Rheum. 2010;62(2):542–552.
- Shirota Y, Yarboro C, Fischer R, et al. Impact of anti-interleukin-6 receptor blockade on circulating T and B cell subsets in patients with systemic lupus erythematosus. Ann Rheum Dis. 2013;72(1):118–128.
- Araki M, Matsuoka T, Miyamoto K, et al. Efficacy of the anti-IL-6 receptor antibody tocilizumab in neuromyelitis optica: a pilot study. Neurology. 2014;82(15):1302–1306.
- Schrezenmeier E, Jayne D, Dörner T. Targeting B cells and plasma cells in glomerular diseases: translational perspectives. J Am Soc Nephrol. 2018;29:741–758.
- Cambridge G, Perry HC, Nogueira L, et al. The effect of B-cell depletion therapy on serological evidence of B-cell and plasmablast activation in patients with rheumatoid arthritis over multiple cycles of rituximab treatment. J Autoimmun. 2014;50:67–76.
- Adlowitz DG, Barnard J, Biear JN, et al. Expansion of activated peripheral blood memory B cells in rheumatoid arthritis, impact of B cell depletion therapy, and biomarkers of response. PLoS One. 2015;10(6):e0128269.
- Romeo AR, Segal BM. Treatment of neuromyelitis optica spectrum disorders. Curr Opin Rheumatol. 2019;31(3):250–255.
- Moreno MJ, Bosch R, Dieguez-Gonzalez R, et al. CXCR4 expression enhances diffuse large B cell lymphoma dissemination and decreases patient survival. J Pathol. 2015;235(3):445–455.
- Reinholdt L, Laursen MB, Schmitz A, et al. The CXCR4 antagonist plerixafor enhances the effect of rituximab in diffuse large B-cell lymphoma cell lines. Biomark Res. 2016;4:12.
- Mikami S, Nakase H, Yamamoto S, et al. Blockade of CXCL12/CXCR4 axis ameliorates murine experimental colitis. J Pharmacol Exp Ther. 2008;327(2):383–392.