
Abstract
Protein kinase CK2, formally known as casein kinase II, is ubiquitously expressed and highly conserved serine/threonine or tyrosine kinase enzyme that regulates diverse signaling pathways responsible for cellular processes (i.e., cell proliferation and apoptosis) via interactions with over 500 known substrates. The enzyme’s physiological interactions and cellular functions have been widely studied, most notably in the blood and solid malignancies. CK2 has intrinsic role in carcinogenesis as overexpression of CK2 subunits (α, α`, and β) and deregulation of its activity have been linked to various forms of cancers. CK2 also has extrinsic role in cancer stroma or in the tumor microenvironment (TME) including the immune cells. However, very few research studies have focused on extrinsic role of CK2 in regulating immune responses as a therapeutic alternative for cancer. The following review discusses CK2’s regulation of key signaling events [Nuclear factor kappa B (NF-κB), Janus kinase/signal transducer and activators of transcription (JAK/STAT), Hypoxia inducible factor–1alpha (HIF-1α), Cyclooygenase-2 (COX-2), Extracellular signal-regulated kinase/mitogen-activated protein kinase (ERK/MAPK), Notch, Protein kinase B/AKT, Ikaros and Wnt] that can influence the development and function of immune cells in cancer. Potential clinical trials using potent CK2 inhibitors will facilitate and improve the treatment of human malignancies.
1. Introduction
George Burnett and Eugene P. Kennedy were the first scientists discovered protein kinase 2 (CK2) approximately 65 years ago, when trying to evaluate an enzyme capable of the phosphorylation of a protein substrate by ATP [Citation1]. Interestingly, the protein kinase was mistakenly named casein kinase II before it was later realized that caseins are only in vitro substrates; whereas, CK2 was initially detected in an animal model (in vivo). Further evaluation of the enzyme protein, throughout the years, would lead scientists to find that CK2 is composed of 2 catalytic subunits, CK2α and CK2α`, and two regulatory subunits, CK2β, that has been identified in multiple forms [Citation2]. Together, the isoforms create a tetrameric, holoenzyme complex with CK2α and/or CK2α` (42 and 38 kDa, respectively) linked to 2 CK2β subunits (28 kDa). CK2β has been proven to be the core of the protein structure due to its ability to stabilize/modulate CK2α subunits and plays an active role in cellular regulation [Citation3]. Buchou et al. [Citation4] performed in vivo experiments with CK2β knock out (KO) mice and in vitro experiments with embryonic fibroblasts depleted of CK2β to showcase the defects in cell proliferation and embryonic lethality that occur in the absence of the subunit. Similar results were reported with CK2α KO mice [Citation5]. The following studies concluded that CK2β and CK2α play an essential role in the development and differentiation, most notably in vertebrates. CK2α` KO mice, on the other hand, are still viable but produce sterile offspring, indicating its significant function in spermatogenesis [Citation5–7]. Prior studies have even demonstrated that protein kinase CK2 subunits are able to functionally exist outside the confinements of the holoenzyme [Citation8]. The omnipresent enzyme protein undergoes dynamic shuttling between cytoplasmic and nuclear compartments as well as intracellular shuttling, which reinforces the ideology that CK2 plays a critical role in the regulation of diverse cell signaling responses [Citation8–10]. More specifically, CK2 is a critical regulator for cellular development, proliferation, and survival [Citation11]. Prior studies have also reported that the CK2 enzyme protein changes cell morphology, enhances cellular transformation, and promotes angiogenesis [Citation12]. Interestingly, elevated levels of CK2 have been observed in malignant cells and are used to determine prognosis as well as survival in various forms of cancer (i.e., breast, prostate, ovarian, lung, colon, and pancreatic) [Citation12]. Scientists have considered the option of using the kinase protein for targeted cancer therapy being that it is a potent suppressor of cell death (apoptosis), which is one of the hallmarks of cancer. Previous studies have successfully demonstrated inhibiting CK2 expression utilizing peptides, RNAi strategies, and other molecules leading to cancer cell deaths both in vivo (xenograft tumors) and in vitro [Citation13,Citation14]. The following review will evaluate the molecular mechanisms behind the CK2’s intrinsic role in solid tumors as well as in blood cancer. CK2’s extrinsic role to influence the immune cell development and function in the TME along with its ability to regulate inflammatory, oncogenic signaling pathways will also be explored. This comprehensive review of CK2 and its relevance to hematological and solid cancers may help lead to the development of new therapeutic regimen.
2. Influence of CK2 expression on cancer cells
Since CK2 is expressed in both normal and cancer cells, it was initially thought to be a marker of cellular proliferation [Citation15,Citation16]. Previous studies have subsequently proved that elevated levels of CK2 in the nucleus as well as its suppression of apoptosis and induction of dysplasia were specific to cancer cells [Citation9,Citation10,Citation17–19]. In fact, upregulated CK2 protein expression has been observed in most of the tumors investigated [Citation16,Citation20], emphasizing its role in tumorigenesis. More specifically, overexpression of CK2 has been detected in breast, prostate, kidney, lung, head and neck, cervical, glioblastoma, ovarian, melanoma, bladder, mesothelioma, endometrium, gastrointestinal (i.e., biliary, liver, esophageal, gastric, colorectal, pancreatic and cholangiocarcinoma), as well as in blood cancers (i.e., myelomas, leukemias and lymphomas). Mechanistically CK2 regulates the activity of at least 15 cancer-related proteins specifically tumor suppressor TP53, the histone deacetylases HDAC1 and HDAC2, and nuclear factor-kappa B (NF-kB) subunit p65 [Citation21]. The following section will discuss in detail the effects of CK2 expression in solid tumors and blood cancers.
2.1. Solid tumors
Elevated CK2 levels have been detected in the tumor microenvironment (TME) of both animal models and humans. Landesman-Bollag and colleagues [Citation22] reported that dysregulated expression of CK2 is associated with and can contribute to tumorigenesis in the mammary glands of humans, rats, and transgenic mice. The research group also detected elevated CK2 levels in both human and rat breast tumor tissues. More specifically, there was an overexpression of the CK2α subunit in the mammary glands of mice which resulted in hyperplasia and dysplasia with a marked percentage of mice developing spontaneous adenocarcinomas [Citation22]. Overexpression of CK2α in the nucleus of human prostate cancer cells has been associated with unfavorable prognostic outcomes [Citation23] and antisense CK2α caused the induction of apoptosis in a human prostate cancer xenograft mouse model [Citation24]. Elevated CK2 activity was also observed in human renal cell carcinoma samples, which correlated with increased ratios of the α and β subunits in tumor versus normal tissues, at the protein level [Citation25]. Additionally, increased CK2 activity has been detected in both the nuclear and cytosolic compartments of squamous cell carcinoma of the head and neck [Citation26] and is associated with the aggressive nature and clinical outcome of the disease [Citation27]. Similar findings were reported in squamous cell carcinoma of the lung [Citation28] and larynx [Citation29]. In human colorectal cancers, overexpression of CK2α in the nucleus is common and serves as a poor prognostic indicator due to its correlation with tumor invasion, stage, and other critical factors [Citation30]. More recently, studies in glioblastoma multiforme (GBM), the most aggressive form of brain tumor, also showed elevated expression of CK2α [Citation31]. Similar findings have been reported for the β regulatory subunit of CK2 in various malignancies. In gastric carcinoma patients, nuclear CK2β was overexpressed and correlated with poor overall survival rates [Citation30]. CK2β is also frequently expressed in endometrial carcinoma, with significant increases apparent during the menstrual cycle in comparison to normal endometrium tissue [Citation32]. This data supports previous findings of the inhibition of CK2 having anti-tumor effects in a mouse xenograft model of human glioblastoma [Citation33].
CK2 activity has been implicated in the survival and resistance to apoptosis for the solid tumors. A number of in vitro as well as in vivo studies downregulating CK2 activity/expression using selective inhibitors, siRNA, antisense oligodeoxynucleotides, or kinase-inactive mutants of CK2 have shown that cancer cells heavily rely on CK2 activity/expression for survival [Citation34]. This dependency or ‘addiction’ to CK2 for survival is also a common phenomenon in malignancies where increased CK2 activity has not necessarily been reported, including in osteosarcomas, ovarian carcinomas, hepatocellular carcinomas, cervical and pancreatic cancers [Citation35]. More importantly, the increased susceptibility of many cancer cell lines to apoptosis as a result of inhibition of CK2 activity increases sensitivity to chemotherapeutic drugs [Citation35,Citation36]. For example, in human pancreatic cancer, RNA interference against the catalytic subunits of CK2 increases sensitivity to gemcitabine-induced cell death [Citation37]. Considering the fact that chemotherapeutic resistance is an impediment to successfully treating pancreatic and other aggressive cancers, these findings point to the potential of CK2 inhibitors in combination with traditional chemotherapy drugs/chemo-preventive agents as a novel treatment strategy to increase the efficacy of these drugs and susceptibility of cancer cells to apoptosis. The dependency of cancer cells on CK2 may be exploited to specifically target neoplastic versus normal cells.
2.2. Blood cancers
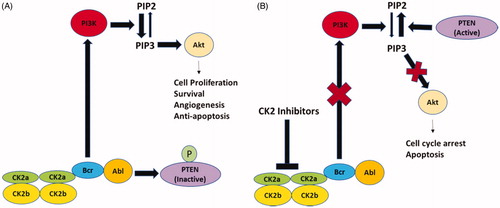
Consistent with studies in solid tumors, the overexpression of CK2 and its regulation of apoptosis also occurs in blood malignancies. Increased CK2 activity and protein expression have been detected in plasma cells of multiple myeloma patients compared to control subjects. In vitro inhibition of CK2 activity had a cytotoxic effect on multiple myeloma plasma cells and increased their sensitivity to the chemotherapeutic agent, melphalan [Citation38]. In fact, CX-4945, a small molecule inhibitor of CK2 [Citation39] has been tested in a Phase I clinical trial in patients that have relapsed, experienced refractory multiple myeloma or have advanced solid tumors. In human T cell acute lymphoblastic leukemia (T-ALL), activation of the PI3K/AKT pathway is attributed to reduced phosphatase activity of PTEN. This occurs when CK2 becomes overexpressed, which causes the Bcr-Abl to activate AKT via the phosphorylation of PIP2 and PIP3 () [Citation40–42] This chain of events then increases cell proliferation, survival, growth, and angiogenesis. Contrastingly, the inhibition of CK2 promotes death within the cell (). Overexpression of CK2α contributed to the development of T cell leukemia and lymphoma [Citation40,Citation42]. Kim et al. [Citation43] studied elevated CK2α expressions in patients with acute myeloid leukemia and found it to be an unfavorable prognostic marker and therapeutic target for inducing cell death. Martins and colleagues [Citation44] found similar results with chronic lymphocytic leukemia patients.
Figure 1. Schematic depiction of protein kinase CK2-mediated signaling in chronic myeloid leukemia. (A) The Bcr moiety of Bcr-Abl interacts with and activates CK2 which then phosphorylates and inactivates PTEN. Bcr-Abl stimulates PI3K activity which catalyzes the conversion of PIP2 to PIP3. PIP3 has a pivotal role in Akt activation leading to enhanced cell proliferation, survival, angiogenesis and anti-apoptosis responses. (B) Treatment with CK2 inhibitors inhibits PI3K activity and PTEN dephosphorylates PIP3 to PIP2 leading to inhibition of Akt activity. Inhibition of Akt activity results in the cell cycle arrest and induction of apoptosis. PI3K-phosphatidylinositol 3-phosphate kinase; PIP2-phosphatidylinositol 4,5-bisphosphate; PIP3-phosphatidylinositol 3,4,5-trisphosphate; PTEN- phosphatase and tensin homolog.
3. Inflammatory and oncogenic signaling molecules regulate immune responses through CK2 expression
As previously highlighted, several studies have investigated the role of CK2 in cancer. Very few of these have focused on CK2’s involvement in regulating immune responses in the tumor microenvironment (TME). It is well understood that CK2 regulates and interacts with oncogenes, tumor suppressors, and other important genes involved in various cellular processes (i.e., transcription, translation, chromatin remodeling, cell cycle progression, proliferation, survival, apoptosis, metastasis, invasion and control of protein stability and degradation). A number of these genes are also involved in key inflammatory signaling pathways that can influence the outcome of immune responses. However, the investigation of CK2’s contribution to these inflammatory signaling pathways, immune responses, and development of cancer is still not quite clear. CK2 regulates several oncogenic signaling pathways that can influence the activation, proliferation, survival, differentiation and function of immune cells. Many of these pathways are deregulated in immune cells and cancers suggesting the possible involvement of CK2 in these altered processes. To investigate this possibility, we summarized key inflammatory and oncogenic signaling molecules regulated by CK2 (), and their specific roles in immune cell development and function ().
Figure 2. Inflammatory and Oncogenic Signaling Pathways regulated (either activated or inhibited) by CK2. CK2 upregulates the expression/activities of inflammatory signaling molecules such as NF-kB, COX-2, JAK/STAT and HIF-1α. CK2 also upregulates the expression/activities of oncogenic signalizing molecules such as AKT, Wnt and ERK. Interestingly CK2 downregulates the expression/activities of Notch and Ikaros as these molecules act as tumor suppressors.
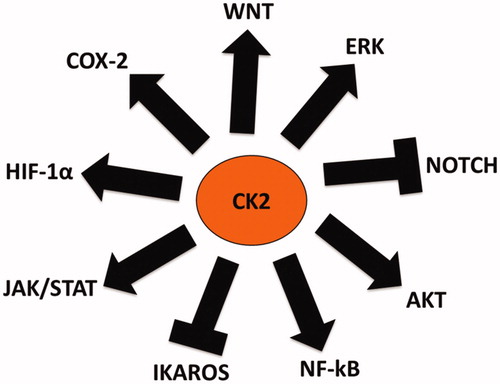
Table 1. Summary of Immune cells whose development and function are regulated by CK2 signaling molecules.
3.1. NF-κB
Nuclear Factor-kappa B (NF-κB) is an inflammatory transcription factor [Citation45]. Several studies have shown that CK2 phosphorylation of IκB leads to IκB degradation and NF-κB activation independent of inhibitors of κB kinase (IKK) activity [Citation46–48]. It activates several gene transcription involved in cellular processes [Citation49], specifically immune cell development, inflammation, proliferation and apoptosis [Citation50,Citation51]. Interestingly, several studies have demonstrated that CK2 downregulates the functionality of NF-kB. Quotti Tubi et al. hypothesized that blocking both CK2 and NF-kB would increase the cytotoxic effect on leukemia stem cells [Citation52]. In addition, CK2 activity results in the activation of NF-κB via its ability to phosphorylate the p65 subunit of the NF-κB dimer in response to IL-1 in hepatocytes as well as in fibroblasts [Citation53] and controls TNF-α dependent phosphorylation of p65 [Citation54]. CK2 phosphorylation and activation of NF-κB is also involved in NF-κB transcription of iNOS in response to IL-1 and LPS [Citation55]. In intestinal epithelial cells, IL-1 stimulation leads to enhanced activation of CK2 bound to IKK or the p65 subunit of the NF-κB and inhibition of CK2 activity attenuated the expression of NF-κB-transcribed proinflammatory factors [Citation56]. Therefore, CK2 regulation of NF-κB could influence its role in immune cells, which may contribute to the development of cancer. The inhibition of NF-κB through CK2 targets tumor not only directly by blocking anti-apoptosis mechanisms of malignant cells, but also indirectly by shifting macrophages from the tumor-tolerating M2-polarization stage towards the tumor-attacking M1-stage [Citation57]. It should be noted that NF-κB also plays a primary role in hematopoiesis via the maturation of hematopoietic stem cells into functional immune cells utilized by both the innate and adaptive immune systems [Citation58]. More specifically, the transcription factor is involved with the survival of dendritic cells and neutrophils during lymphopoiesis to regulate the proliferation and maturation of B and T cells. Additionally, NF-κB activation is important for macrophage viability and function in tumors [Citation59,Citation60]. Adaptive immune responses depend on NF-κB activation for toll-like receptor (TLR) pathway signaling. Proliferating T cells require NF-κB for pro-survival and cytokine production necessary for their proliferation and differentiation. For instance, NF-κB activation is essential for Treg differentiation [Citation61]. It enhances the expression of cytokines (TNF-α, IL-1, IL-6 and IL-8), chemokines [chemokine (C-C motif) ligand 2 (CCL2)], adhesion molecules 1 (VCAM-1) and the Intracellular Cell Adhesion Molecule 1 (ICAM-1)], matrix metalloproteinases (MMPs), cyclooxygenase 2 (COX2), inducible nitric oxide synthase (iNOS) and antiapoptotic protein (B-cell lymphoma 2) [Citation62,Citation63], all of which can influence leukocyte development, function and chemotaxis.
3.2. Janus kinase/signal transducer and activators of transcription (JAK/STAT)
JAK-STAT signaling is involved in the regulation of cell survival, proliferation, and differentiation [Citation64]. JAK tyrosine kinases constitutively activated and phosphorylated STATs leading to regulation of the transcription of inflammatory cytokines as a result of mutations that affect their function in solid tumors and hematopoietic tumors [Citation65–67]. CK2 binds to and phosphorylates JAK2 leading to activation of JAK/STATs signaling and induction of gene expression of proteins involved in growth, anti-apoptotic mechanisms, angiogenesis, and immune evasion of tumor cells [Citation67,Citation68]. The inhibition of CK2 activity prevents cytokine-induced activation of JAK/STATs, inhibits downstream gene expression, and induces apoptosis in human erythroid leukemia (HEL) cells [Citation68,Citation69]. Each STAT family member protein plays a critical role in cytokine-dependent inflammation and immune modulation [Citation67]. In the TME, STAT proteins are the determining factor as to whether immune responses will act to promote or inhibit cancer progression via pathways regulating inflammation [Citation67]. There is a cross talk between STAT3 and NF-kB and both cooperatively regulate anti-apoptotic, cell cycle, cytokines and chemokines genes in TME [Citation70]. It has been shown that NF-κB interact physically with STAT3, facilitating NF-κB recruitment to STAT3 promoters and vice versa [Citation70]. JAK3 is highly expressed in hematopoietic cells and is required for IL-2, IL-4, IL-7-, IL-9, IL-15 and IL-21 cytokine signaling; meanwhile, the STAT1 isoform is necessary for IFN signaling. These cytokines are critical mediators of lymphocyte development and function. JAK/STAT pathways play an important role in T cell lineage fates, particularly Th1 versus Th2. IL-4 stimulation causes STAT6 activation and the generation of IL-4 secreting Th2 cells, while IL-12 induces STAT4 activation resulting in IFN-γ-secreting Th1 cells [Citation71,Citation72]. In Tregs, STAT5 plays an important role in their development and function [Citation73,Citation74] and STAT3 regulates the generation of Th17 cells [Citation74]. Furthermore, STAT3 is critical in generating effector B cells [Citation75]. In dendritic cells, JAK3 is vital for their maturation, migration and function [Citation76]. JAKS also modulate macrophage production of inflammatory cytokines [Citation77]. STATs have been found to promote cellular proliferation and apoptosis resistance in human multiple myeloma cells and promote tumor tolerance by immune cells [Citation78]. Since CK2 inhibition is associated with the impairment of STAT3 activation and signaling, a rational use of CK2 inhibitors would be together with STAT3 inhibitors. Therefore, CK2 and STAT3 inhibitors may cooperate in inducing antitumor immune responses. The combination of CK2 inhibitors with STAT3 inhibitors would likely to have synergistic antitumor activity and more efficacious for cancer immunotherapy.
3.3. HIF-1α
Hypoxia Inducible Factor (HIF) is a central transcription factor controls the cellular adaptation to a lack of oxygen or hypoxia [Citation79]. Hypoxia occurs in most solid tumors due to inefficient vasculature and contributes to resistances observed in chemotherapy as well as radiation therapy [Citation79,Citation80]. CK2 activity and levels are elevated in hepatocellular and cervical cancer cells grown under hypoxia, concomitant with increased nuclear localization [Citation81,Citation82]. CK2 phosphorylates HIF-1α and regulates its activity [Citation81]. Hubert et al. [Citation79], showed that CK2 may play a role in regulating HIF-1α as inhibition of CK2 activity resulted in decreased HIF-1α activity under hypoxic conditions through an increase in p53, a well-known CK2 target. This transcription factor potentially mediates inflammation via its regulation of hypoxia induction of NF-κB and its NF-κB-dependent activation by proinflammatory TNFα and IL-1β, in normoxic and cancer cells [Citation83–85]. Changes in metabolism during inflammation can result in significant tissue hypoxia and thus, the induction of HIF-1α as well as hypoxia-responsive genes [Citation86]. Direct transcriptional up-regulation of cyclooxygenase-2 by hypoxia-inducible factor (HIF)-1 promotes colorectal tumor cell survival and enhances HIF-1 transcriptional activity during hypoxia [Citation87]. Mirzoeva et al. reported that CK2 inhibitor (Apigenin) inhibits hypoxia by disrupting the interaction between HIF-1α protein and HSP90 leading to suppression of prostate cancer angiogenesis [Citation88]. Additionally, HIF-1α mediates leukocyte adhesion and myeloid cell infiltration at inflammatory sites [Citation89,Citation90]. When cells experience a deficiency in HIF-1α, it limits the development of B cells in the bone marrow and plays a crucial role for activating dendritic cells in inflammatory conditions [Citation91,Citation92]. HIF-1α also limits CD4+ and CD8+ T cell production of TNFα and IFNδ and their proliferation after T-cell receptor activation [Citation93]. Our laboratory demonstrated that CK2 inhibitor (Apigenin) improves T cell homeostasis and function in murine pancreatic cancer [Citation94]. Therefore, the association between HIF-1α, inflammation, immune modulation, and its regulation by CK2 should be further investigated.
3.4. COX-2
Cyclooxygenases-2 (COX-2) is a prostaglandin (PG)–endoperoxide synthase 2 enzyme responsible for generation of prostaglandin E2 (PGE2) that is contributed to the modulation of pro-carcinogenic effects [Citation95,Citation96]. CK2 up‐regulates COX‐2 expression by modulating the Wnt/β‐catenin signaling pathway and promotes the viability of human embryonic kidney cells [Citation97]. Subsequent studies showed that CK2 was also a positive regulator of COX-2 dependent of PGE2 production, which correlated with increased viability of human colorectal and breast cancer cells [Citation97]. CK2‐dependent up‐regulation of COX‐2 expression is linked to the expression of cancer‐related genes (AKT, β‐catenin and survivin) as COX-2 is also a target of the Wnt/β-catenin pathway, a well-characterized substrate of CK2 [Citation98]. Thus, CK2 inhibitors would be useful to inhibit COX-2 and oncogenic pathway leading to regression of tumor growth. Additionally, COX‐2 is released by cancer‐associated fibroblasts (CAFs), macrophage type 2 (M2) cells, Tregs and cancer cells to TME [Citation96] and COX‐2 mediated hypoxia within the TME along with its positive interactions with YAP1 and antiapoptotic mediators are responsible for cancer cell resistance to chemotherapeutic drugs [Citation96,Citation99,Citation100]. Furthermore, enhanced COX-2 expression and its product PGE2 have been linked to immunosuppression in cancers due to the associated increase in IL-10, decrease in IL-12, IL-1 and TNFα [Citation57,Citation96,Citation101,Citation102] as well as development of immunosuppressive MDSC and the induction of Tregs [Citation103,Citation104]. In CD8+ T cells, PGE2 accelerates their progression towards replicative senescence and their associated effector functions [Citation96,Citation105]. COX‐inhibitors relieve the immunosuppressive effect of tumor cells and improve functions of immune effectors [Citation99,Citation100,Citation106]. In summary, CK2 plays an important role in regulating COX-2 production of PGE2 in the TME due to its involvement in inflammation, immune cells and tumorigenesis. Therefore, the combination of CK2 inhibitors with COX-2 inhibitors would likely to have synergistic activity and more efficacious for cancer chemotherapy. Additional studies need to be done to confirm/elaborate this theory.
3.5. ERK/MAPK
Extracellular Signal-Regulated Kinase (ERK) is a member of the mitogen-activated protein kinase (MAPK) family downstream of the activated oncogenic RAS GTPase and activating mutations in these genes lead to oncogenesis and immune responses [Citation106–109]. The Ras/Raf/MEK/ERK pathway is constitutively active in several cancers due to mutations in oncogenic RAS and signaling molecules [Citation110]. CK2 is primarily responsible for phosphorylating ERK at its nuclear translocation signal allowing its nuclear import [Citation111] and binding to transcription factors that induce gene expression cell proliferation, differentiation, apoptosis and tumorigenesis [Citation112]. Elevated ERK expression has been detected in various human tumors, such as ovarian, colon, breast, lung and blood cancer [Citation111,Citation113–115]. The inhibition of CK2 is particularly potent in inducing apoptosis in HEL cells, which is associated with suppression of constitutive activation of ERK [Citation111]. Additionally, ERK regulates the production of TNF-α and macrophage function [Citation116]. The Activation of the ERK pathway is critical for T cell activation as well as in IL-2 expression and the differentiation of CD4+ T helper 2 (Th2) cells [Citation117,Citation118]. In dendritic cells, ERK regulates this suppressive function through Fas signaling [Citation119]. ERK signaling also plays a role in B cell differentiation as well as macrophage differentiation and polarization [Citation107,Citation120]. The interaction between ERK and CK2 may therefore be important in regulating immune responses. Since CK2 inhibition is associated with the impairment of ERK activation and signaling, a rational use of CK2 inhibitors would be together with ERK inhibitors for synergistic effects.
3.6. Notch
Notch signaling pathway plays a vital role in transmitting and converting extracellular ligand signals into transcriptional responses in the nucleus [Citation121]. CK2 has been demonstrated to be a regulator of the Notch pathway through the phosphorylation of Notch intracellular domain (NICD). Ranganathan et al. have identified multiple CK2 phosphorylation sites, located in the ankyrin domain of Notch. Phosphorylation of both sites resulted in decreased binding of NICD to DNA and consequently lower transcriptional activity [Citation122]. Recently, researchers have established a relationship between the Notch signaling pathway and CK2 expression [Citation123]. Notch1 protein levels were reduced after CK2α expression was silenced in cancer cells [Citation123]. In addition, inhibition of CK2 using a small molecule inhibitor decreased Notch1 transcriptional activity and reduced the stem-cell like CD44+/CD24-cell population [Citation124,Citation125]. Notch activity, which inhibits the differentiation of MDSCs from hematopoietic stem cells and common myeloid progenitors, decreases with elevated levels of CK2 expression, a phenotype observed in cancer patients. This causes MDSCs, both monocytic and granulocytic, to accumulate in TME [Citation126]. Cheng and colleagues [Citation124] found that MDSCs from EL4 TB mice that have been transfected with control siRNA and cultured in tumor cell conditioned medium (TCM) had substantially lower Notch signaling (as measured by Hes1 expression) compared to cells cultured in complete medium [Citation127]. Similar results were observed for MDSCs transfected with CK2 siRNA and cultured with TCM. The study concluded that CK2 has a negative impact on Notch signaling, which contributes to increases in MDSC differentiation in the TME. Notch signaling is essential for T cell precursor and B cell development and differentiation. Thymus settling progenitors develop into several states of CD4– and CD8– double negative T progenitor cells that eventually differentiate into T cells with increased Notch activity [Citation128]. Interestingly, Notch signaling is more likely to produce γδ T cells in humans and αβ T cells in mice. Previous studies have also shown that Notch activity promotes the differentiation of Th1, Th2, Th9, and Th17 through the up regulation of molecules such as SMAD3, Tbx2, and IL23r [Citation128,Citation129]. Notch signaling is required for T cell precursor and B cell development and differentiation. Notch also plays a role in dendritic cell development and maturation and myelopoiesis with altered Notch signaling contributing to MDSC development [Citation129]. Notch signaling is also important for TCR-activation and peripheral T cell proliferation and the cytotoxic abilities of CD8+ T cells mediated by perforin and granzyme B expression. In addition, Notch activation regulates the differentiation of CD4+ T cells to Th1 vs Th2 cells by regulating the expression of key transcription factors and cytokines. Studies have also shown that Notch regulates CD4+ and CD8+ T cell cytokine production of IL-10 and IFN-γ and IL-17, respectively [Citation130]. Increased CK2 activity and phosphorylation is also proposed to downregulate Notch signaling in hematopoietic progenitor cells and MDSC, emphasizing its role in myeloid cell differentiation [Citation127]. CK2 downregulates Notch signaling in cancer cells and reduces a stem-cell like population in these cells [Citation123–125]. Therefore, CK2 inhibitors are useful in combating the cancer progression by regulating the Notch pathway.
3.7. Protein kinase B/AKT
Protein Kinase B (PKB or AKT), a serine/threonine kinase has diverse roles in cell proliferation, growth, survival, metabolism, angiogenesis, cancer, and immunity [Citation131]. AKT is most frequently activated and overexpressed in solid tumors as well as in hematological malignancies and referred as oncogenes [Citation132]. CK2 has been shown to phosphorylate Akt at Ser 129 leading to its activation of downstream signaling events in various cell types [Citation133]. Furthermore, the interaction between CK2 subunits and AKT enhance AKT’s activity [Citation134,Citation135]. Ruzzene and colleagues [Citation135] demonstrated the relationship between CK2 and AKT. CK2 and AKT share global pro-survival and anti-apoptotic functions. Their activities are aberrantly high in human cancers, and their signaling pathways potentiate each other by several levels of cross-talk. Both α and β CK2 intervene in the direct binding to AKT resulting in an increased AKT activity. AKT signaling effects are mediated in conjunction with phosphatidylinositol 3-kinase (PI3K) and mammalian target of rapamycin (mTOR). This PI3K/AKT/mTOR pathway is posed to be the most frequently activated pathway in human cancers with an occurrence rate of 30–50% [Citation136]. Additionally, The PI3K-AKT pathway regulates both myeloid cells of the innate immune system and lymphoid cells of the adaptive immune system. In myeloid cells, AKT is activated by growth factors, cytokines (e.g., IL-4), and ligands for toll-like receptors (TLRs; e.g., LPS) and AKT activation stimulates a similar array of downstream effectors across the myeloid lineage [Citation131]. AKT mediates macrophage polarization in response to different stimuli. M1 state is promoted by LPS, whereas the M2 state is induced by IL-4. Both M1 and M2 polarizing signals activate AKT and AKT signaling promotes M2 macrophage polarization [Citation137]. In B and T cells, PI3K and mTOR regulate their cell survival, proliferation and B and T cell receptor signaling leading to their activation [Citation138,Citation139]. Similar regulation of survival and differentiation were found in dendritic cells [Citation140]. In macrophages, AKT regulates inflammatory gene expression, their phagocytosis function as well as their migration and chemotaxis [Citation141]. The PI3K/AKT pathway also controls the proliferation, survival, migration and metabolism of T cells [Citation142]. AKT signaling also regulates the differentiation of effector and memory CD8 T cells [Citation143]. The AKT/mTOR signaling pathway also modulates the differentiation of Th17 cells [Citation144]. In regulatory T cells, the PI3K/AKT pathway is necessary for their proliferation and the inhibition of this pathway leads to their depletion in vivo [Citation142]. Our group has published that MDSC have hyperactivation of AKT signaling, which may contribute to their expansion in pancreatic cancer [Citation145]. Both CK2 and AKT phosphorylate each other leading to cellular synergistic effects. Therefore, both are attractive targets for therapeutic intervention of cancer,
3.8. Ikaros
Ikaros is a unique family of zinc finger transcription factors regulates normal hematopoiesis and is required for all lymphoid lineage development in which is critical for a functional immune system [Citation146,Citation147]. Ikaros knock-out mice lack B and T lymphocytes and natural killer (NK) cells, as well as their defined progenitors [Citation146]. Scientists have discovered that CK2 phosphorylates Ikaros in T cells, decreases its DNA-binding affinity for its target genes and re-distributes Ikaros protein in the nucleus from pericentromeric localization to a diffuse nuclear-staining pattern [Citation148]. Lack of dephosphorylation of Ikaros by protein phosphatase 1 (PP1) leads to increased proteasomal degradation of Ikaros due to protein kinase CK2-mediated hyperphosphorylation at PEST regions [Citation149]. Phosphorylation of Ikaros by CK2 regulates Ikaros binding and repression of the terminal deoxytransferase (TdT) gene in normal thymocytes and in T-cell ALL [Citation150]. Overall, increased CK2 activity is thought to impede and/or downregulate Ikaros’ function, causing malignant transformation [Citation151]. Cho et al. investigated the possible involvement of CK2-regulation of Ikaros in inflammation by showing Ikaros phosphorylation by CK2 controls Ikaros’ regulation of iNOS in response to LPS/IFN-γ stimulation in macrophages [Citation152]. Furthermore, our recent findings also suggest a negative role for CK2 in regulating Ikaros stability in T cells to maintain effector and regulatory T cell homeostasis in murine pancreatic cancer [Citation94]. Pancreatic tumor-derived factors (TDF) enhanced CK2 activity and inhibited protein phosphatase 1 (PP1) activity. CK2 hyperphosphorylated Ikaros leading to polyubiquitination and degradation by the ubiquitin-proteasome system and loss of T cell homeostasis in pancreatic tumor (, modified from Nelson study) [Citation94]. CK2 inhibitor apigenin (API) inhibited CK2 activity and stabilized Ikaros expression by increasing PP1 activity resulting in T cell homeostasis and anti-tumor immune response. Proteasome inhibitor MG132 directly inhibited activity of the ubiquitin-proteasome system and prevented the proteasomal degradation of Ikaros and thereby stabilized its expression (, modified from Nelson study) [Citation94]. Considering the evidence summarized in this review, it is possible that increased CK2 activity may be a result of proinflammatory stimuli affecting the downstream functions of Ikaros as a tumor suppressor. Thus, leading to tumorigenesis and deregulated expression of Ikaros-modulated inflammatory cytokines and genes; essentially resulting in altered immune cell development and function, further contributing to the observed cancer cell phenotypes.
Figure 3. Proposed model of Ikaros' regulation by CK2 in pancreatic tumor. (A) Pancreatic tumor-derived factors. (TDF) enhanced CK2 activity and inhibited protein phosphatase 1 (PP1) activity. Enhanced CK2 activity hyperphosphorylated Ikaros leading to polyubiquitination and protein degradation by the ubiquitin-proteasome system. (B) CK2 inhibitor Apigenin (API) inhibited CK2 activity and stabilized Ikaros expression by increasing PP1 activity which resulted in T cell homeostasis and anti-tumor immune responses. Proteasome inhibitor MG132 in vitro directly inhibited the activity of the ubiquitin-proteasome system, prevented proteasomal degradation of Ikaros and stabilized its expression in pancreatic tumor microenvironment. Modified from Nelson study [Citation94].
![Figure 3. Proposed model of Ikaros' regulation by CK2 in pancreatic tumor. (A) Pancreatic tumor-derived factors. (TDF) enhanced CK2 activity and inhibited protein phosphatase 1 (PP1) activity. Enhanced CK2 activity hyperphosphorylated Ikaros leading to polyubiquitination and protein degradation by the ubiquitin-proteasome system. (B) CK2 inhibitor Apigenin (API) inhibited CK2 activity and stabilized Ikaros expression by increasing PP1 activity which resulted in T cell homeostasis and anti-tumor immune responses. Proteasome inhibitor MG132 in vitro directly inhibited the activity of the ubiquitin-proteasome system, prevented proteasomal degradation of Ikaros and stabilized its expression in pancreatic tumor microenvironment. Modified from Nelson study [Citation94].](/cms/asset/7b9d9463-3a9a-4728-87ac-e8bd748d8688/timm_a_1843267_f0003_c.jpg)
3.9. Wnt
Wnt known as ‘Wingless-related integration site’ is a family of secreted cysteine-rich glycoproteins regulates cell proliferation, survival and fate determination [Citation153]. CK2 function is required in Wnt producing cells for Wnt secretion [Citation154]. CK2 is a positive regulator of Wnt pathway at multiple levels [Citation155]. It phosphorylates intracellular Wnt signaling intermediates Dvl, β-catenin, and T cell-specific transcription factor/lymphoid enhancer-binding factor (TCF/LEF) [Citation156]. Wnt signaling regulates the tumor-immune cycle in dendritic cells, T cells, and tumor cells. Increasing evidence suggests that Wnt signaling blocks the tumor-immune cycle at all steps, including tumor antigen release, antigen presentation, T cell priming and activation, T cell infiltration, and tumor cell elimination [Citation157]. In the TME, activation of β-catenin depletes chemokine CXCL9/10 in CD103+ DCs and inhibits infiltration of effector T cells [Citation158]. In addition, abnormal activation of Wnt signaling in DCs disrupts cross-priming of T cells by increasing the concentration of IL-10 [Citation159]. Inactivation of Wnt signaling by deletion of Wnt receptor’s low-density lipoprotein receptor-related protein 5/6 (LRP5/6) in DCs facilitates tumor antigen capture and cross-priming, upregulates proinflammatory cytokines, and downregulates immune regulatory factors (IL-10 and TGF-β1), and eventually promotes T cell antitumor immune response [Citation160]. Abnormal Wnt signaling directly alters several regulators critical for the antitumor activities of T cells, especially effector T cells, T helper cells, and regulatory T cells. Wnt signaling and TCF1 are required for generation of memory CD8+ T cells as well as generation and activation of CD8+ effector T cells [Citation161]. Wnt signaling inhibits T helper (Th) cells and impairs antitumor immunity as well as the immunity of CD4+ T cells [Citation162]. Wnt signaling, one of the best-characterized cancer drivers, have been tightly connected to the initiation and progression of many cancer type [Citation158]. CK2-induced mammary tumors have upregulated nuclear β-catenin and CK2 inhibitors reduced proliferation and β-catenin expression in tumor cells [Citation163]. Wnt signaling significantly activated in colorectal cancer and the antitumor immunity of CD4+ T cells is weakened by repressing IFN-γ expression and elevating IL-17a expression which can be involved in both pro-tumorigenic activity and in antitumor immunity [Citation162]. Therefore, targeting Wnt signaling by CK2 inhibitors would potentially improve clinical outcomes of cancer patients by overcoming the primary, adaptive, and acquired resistance to immunotherapy.
3.10. CK2 inhibitors
With studies providing evidence that CK2 plays a critical role in cellular processes that lead to tumorigenesis, it is no surprise that emerging research has extensively focused on creating specific inhibitors of CK2. The CK2 protein’s ATP binding site contains unique bulky residues that favor the design of ATP-competitive inhibitors [Citation164]. The most commonly studied inhibitors are CX-4945 [(5-(3-chlorophenyl)amino)-benzo(c)-2,6-naphthyridine-8-carboxylic acid)], CIGB-300, TBB (4,5,6,7-tetrabromo-1H- benzotriazole), DMAT (2-dimethylamino-4,5,6,7-tetrabromo- 1H-benzimidazole), Quinalizarin, hematein, TBCA (Tetra bromo cinnamic acid), DRB [5,6-di-chloro-1-(b-D-ribofuranosyl)-benzimidazole], Apigenin (4′,5,7,-trihydroxyflavone), Emodin (1,3,8-trihydroxy-6-methyl-antraquinone) and TF (6,7-dichloro-1,4-dihydro-8-hydroxy-4-[(4-methylphenylamino) methylene] dibenzo [b, d] furan-3(2H)-one). DRB, one of the first identified CK2 inhibitors, lacks the ability to inhibit other kinases with similar efficacy to CK2 [Citation165,Citation166]. Flavonoids, natural compounds found in fruit, vegetables, wine and tea, have also demonstrated the ability to inhibit CK2 activity. Two primary examples of flavonoids used for CK2 inhibition include apigenin and quercetin, both of which are more potent inhibitors of CK2 activity than DRB but maintain limited specificity [Citation167]. Interestingly, apigenin is now characterized as a selective CK2 inhibitor with pro-apoptotic, anti-inflammatory, anti-cancer and chemo preventative effects. Another natural product shown to inhibit CK2 and other kinases with a broad specificity is emodin [Citation168]. Currently, the most commercially available CK2 inhibitors are TBB and DMAT are specific to CK2 [Citation164]. TBB is a derivative of DRB while DMAT is a derivative of TBB’s analogue, TBI (Tetra Bromo-benzimidazole) [Citation164,Citation169,Citation170]. IQA ([5-oxo-5,6-dihydro-indolo(1,2-a) quinazolin-7-yl] acetic acid), another CK2 inhibitor, shows enhanced selectivity and efficacy in vitro and in vivo compared to TBB, emodin and apigenin [Citation167]. More recently, Cylene Pharmaceuticals developed the first orally bioavailable CK2 inhibitor called CX-4945 also known as Silmitasertib. This potent and highly selective, small molecule inhibitor blocks both CK2α and CK2α’ while causing cell cycle arrest, inhibition of cellular proliferation pathways and the promotion of apoptosis [Citation171]. Interestingly, CX-4945 advanced to clinical trials in patients with solid tumors. Clinicians showed that the drug was well-tolerated with minimal side effects, inhibited CK2 and AKT pathways, caused a reduction in circulating tumor cells and IL-6 and IL-8 and stabilized disease in some patients [Citation39]. CX-4945 also sensitized AML cells to daunorubicin (AML chemotherapeutics) and induced apoptosis of T-ALL cells by down-regulated PI3K/Akt/mTOR signaling pathway [Citation40,Citation172]. The second CK2 inhibitor known to have entered clinical trials (phases 1 and 2) is CIGB-300, a permeable cyclic peptide known to block CK2 phosphorylation by targeting specific phosphor acceptor domains [Citation173]. Pre-clinical studies have also shown that Human H125 lung carcinoma cell lines treated with 20 µM and 50 µM of CIGB-300 displayed significant reductions in tumor cell secreting enzymes, urokinase plasminogen activator and matrix metalloproteinase-2 [Citation174]. CIGB-300 decreases cell viability and proliferation in CLL cell lines, enhances apoptosis in primary leukemia cells, and showed anti-cancer activity in a human CLL xenograft mouse model [Citation175]. CK2 inhibitors (TBB and CX-4945) demonstrated a potent therapeutic effect in a panel of patient-derived primary high-risk B-cell acute lymphoblastic leukemia xenografts through prolonged survival and reduction of leukemia burden [Citation151]. New CK2 inhibitors from Bristol Myers Squib company (BMS-211 and BMS-595) showed reduction in the number of PMN-MDSC and TAM by blocking of their differentiation in LLC lung, CT26 colon, 4T1 breast tumor-bearing mouse models [Citation176]. Due to its positive response both preclinically and clinically, CIGB-300 is currently undergoing phase 3 for clinical trials most notably with patients involved with locally advanced cervical cancer. However, caution should be taken while using CK2 inhibitors in some cases. The clinical application of chemical inhibitors is not always right strategy to target CK2 as CK2 functions, which are not dependent on its catalytic activity, but regulatory activity has also been reported [Citation177,Citation178]. The combination of CK2 inhibitors with other drugs might not be efficacious. In cases of melanoma and thyroid carcinoma with WT BRAF, the efficacy of CK2 inhibitors in combination with BRAF/MEK inhibitors was poor [Citation179]. Evidences also support a positive role of CK2 in allowing the cellular response to topoisomerase I-targeting drugs, in this case the combination therapy with CK2 inhibitors would be contraindicated [Citation180,Citation181]. Moreover, whether responsiveness to CK2 inhibition requires p53 functions, which would imply the inadequacy of CK2 targeting in case of TP53 mutation/deletion [Citation31,Citation172,Citation182]. All these observations suggest that the clinical application of CK2 inhibitors should be carefully designed. CK2 is dispensable in the adult organism and in terminally differentiated cells, as inhibition of CK2 activity is not detrimental for the survival of normal cells [Citation183]. CK2 inhibitor has been reported to inhibit the proliferation of chronic lymphocytic leukemia (CLL) cells without affecting the normal T and B cells [Citation44,Citation175,Citation183]. Most importantly, the inhibition of CK2 may inhibit the self-renewal of the stem cell niche, which are also present in different tissues and organs of the adult organism and repair capacity of an organism. In summary, CK2 inhibitors are highly effective against solid tumors (breast, lung, head and neck, cervical, brain, ovarian, liver, and pancreas) and in combination with other therapies for hematological malignancies (Multiple Myeloma, Leukemia and Non-Hodgkin’s Lymphoma). Current and future clinical trials using CK2 inhibitors will be useful in combating this devastating disease in humans.
4. Conclusion
Regulation of signaling pathways that contribute to the development and function of immune cells is an area that requires further investigation. Our understanding as to the involvement of the diverse pathways in these processes as well as how they are connected, remains to be unraveled. CK2 has a clear, intrinsic role in providing a phenotypic advantage to cancer cells resulting in malignant transformation. However, in this review, we have summarized several findings that point to the possible extrinsic role of CK2 in regulating immune cell development and function via its regulation of inflammatory and oncogenic signaling pathways in cancer. Many of these pathways are tightly regulated during immune cell development. Therefore, alterations in their signaling mechanisms in tumor microenvironments can lead to diminished immune responses. Even with this understanding, targeting these signaling molecules with redundant and overlapping functions is a difficult task. Pursuing upstream regulators, such as CK2, are necessary for critical cellular functions, and can prove to be beneficial in controlling these molecular pathways. Furthermore, promising findings highlighted in this review provides evidence for the importance of developing highly specific and effective CK2 inhibitors. Overall, CK2 may serve as a potential therapeutic target for not only targeting tumors but also modulating inflammatory and oncogenic signaling pathways that influence immune responses. The omnipresence CK2’s involvement in diverse cellular processes makes it an attractive anti-therapeutic for restoring immune responses in solid and blood cancers. Potential future studies will hopefully provide an essential piece to the puzzle to our understanding of inflammation and its association with cancer as well as immunotherapeutic responses in preclinical and clinical investigations.
Author contributions
K.H., T.T.W., N.N., and T.G. conceived, planned, and wrote the review article.
Consent for publication
We consent to publication
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability
We agree to share all the data and content of this review.
References
- Burnett G, Kennedy EP. The enzymatic phosphorylation of proteins. J Biol Chem. 1954;211(2):969–980.
- Litchfield DW. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J. 2003;369(Pt 1):1–15.
- Graham KC, Litchfield DW. The regulatory beta subunit of protein kinase CK2 mediates formation of tetrameric CK2 complexes. J Biol Chem. 2000;275(7):5003–5010.
- Buchou T, Vernet M, Blond O, et al. Disruption of the regulatory beta subunit of protein kinase CK2 in mice leads to a cell-autonomous defect and early embryonic lethality. Mol Cell Biol. 2003;23(3):908–915.
- Lou DY, Dominguez I, Toselli P, et al. The alpha catalytic subunit of protein kinase CK2 is required for mouse embryonic development. Mol Cell Biol. 2008;28(1):131–139.
- Escalier D, Silvius D, Xu X. Spermatogenesis of mice lacking CK2alpha’: failure of germ cell survival and characteristic modifications of the spermatid nucleus. Mol Reprod Dev. 2003;66(2):190–201.
- Wirkner U, Pyerin W. CK2alpha loci in the human genome: structure and transcriptional activity. Mol Cell Biochem. 1999;191(1–2):59–64.
- Bibby AC, Litchfield DW. The multiple personalities of the regulatory subunit of protein kinase CK2: CK2 dependent and CK2 independent roles reveal a secret identity for CK2beta. Int J Biol Sci. 2005;1(2):67–79.
- Ahmad KA, Wang G, Unger G, et al. Protein kinase CK2-a key suppressor of apoptosis. Adv Enzyme Regul. 2008;48:179–187.
- Ahmed K, Yenice S, Davis A, et al. Association of casein kinase 2 with nuclear chromatin in relation to androgenic regulation of rat prostate. Proc Natl Acad Sci USA. 1993;90(10):4426–4430.
- Gotz C, Montenarh M. Protein kinase CK2 in development and differentiation. Biomed Rep. 2017;6(2):127–133.
- Chua M, Ortega C, Sheikh A, et al. CK2 in cancer: cellular and biochemical mechanisms and potential therapeutic target. Pharmaceuticals, 2017;10(4):18.
- Ahmed K, Kren BT, Abedin MJ, et al. CK2 targeted RNAi therapeutic delivered via malignant cell-directed tenfibgen nanocapsule: dose and molecular mechanisms of response in xenograft prostate tumors. Oncotarget. 2016;7(38):61789–61805.
- Solares AM, Santana A, Baladrón I, et al. Safety and preliminary efficacy data of a novel casein kinase 2 (CK2) peptide inhibitor administered intralesionally at four dose levels in patients with cervical malignancies. BMC Cancer. 2009;9:146.
- Litchfield DW, Luscher B. Casein kinase II in signal transduction and cell cycle regulation. Mol Cell Biochem. 1993;127–128:187–199.
- Trembley JH, Wang G, Unger G, et al. Protein kinase CK2 in health and disease: CK2: a key player in cancer biology. Cell Mol Life Sci. 2009;66(11–12):1858–1867.
- Guo C, Yu S, Davis AT, et al. A potential role of nuclear matrix-associated protein kinase CK2 in protection against drug-induced apoptosis in cancer cells. J Biol Chem. 2001;276(8):5992–5999.
- Li Q, Zong Y, Li K, et al. Involvement of endothelial CK2 in the radiation induced perivascular resistant niche (PVRN) and the induction of radioresistance for non-small cell lung cancer (NSCLC) cells. Biol Res. 2019;52(1):22.
- Luo W, Yu W-D, Ma Y, et al. Inhibition of protein kinase CK2 reduces Cyp24a1 expression and enhances 1,25-dihydroxyvitamin D(3) antitumor activity in human prostate cancer cells. Cancer Res. 2013;73(7):2289–2297.
- Tawfic S, Yu S, Wang H, et al. Protein kinase CK2 signal in neoplasia. Histol Histopathol. 2001;16(2):573–582.
- Nunez de Villavicencio-Diaz T, Rabalski AJ, Litchfield DW. Protein kinase CK2: intricate relationships within regulatory cellular networks. Pharmaceuticals. 2017;10(4):27.
- Landesman-Bollag E, Song DH, Romieu-Mourez R, et al. Protein kinase CK2: signaling and tumorigenesis in the mammary gland. Mol Cell Biochem. 2001;227(1–2):153–165.
- Laramas M, Pasquier D, Filhol O, et al. Nuclear localization of protein kinase CK2 catalytic subunit (CK2alpha) is associated with poor prognostic factors in human prostate cancer. Eur J Cancer. 2007;43(5):928–934.
- Slaton JW, Unger GM, Sloper DT, et al. Induction of apoptosis by antisense CK2 in human prostate cancer xenograft model. Mol Cancer Res. 2004;2(12):712–721.
- Stalter G, Siemer S, Becht E, et al. Asymmetric expression of protein kinase CK2 subunits in human kidney tumors. Biochem Biophys Res Commun. 1994;202(1):141–147.
- Faust RA, Gapany M, Tristani P, et al. Elevated protein kinase CK2 activity in chromatin of head and neck tumors: association with malignant transformation. Cancer Lett. 1996;101(1):31–35.
- Gapany M, Faust RA, Tawfic S, et al. Association of elevated protein kinase CK2 activity with aggressive behavior of squamous cell carcinoma of the head and neck. Mol Med. 1995;1(6):659–666.
- O-charoenrat P, Rusch V, Talbot SG, et al. Casein kinase II alpha subunit and C1-inhibitor are independent predictors of outcome in patients with squamous cell carcinoma of the lung. Clin Cancer Res. 2004;10(17):5792–5803.
- Wu L, Liu B. [The expressions and significance of CK2 in normal laryngeal mucosa, laryngeal precancerosis and laryngeal squamous cell carcinoma]. Lin Chung Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 2007;21(18):825–827, 830.
- Lin K-Y, Fang C-L, Chen Y, et al. Overexpression of nuclear protein kinase CK2 Beta subunit and prognosis in human gastric carcinoma. Ann Surg Oncol. 2010;17(6):1695–1702.
- Dixit D, Sharma V, Ghosh S, et al. Inhibition of Casein kinase-2 induces p53-dependent cell cycle arrest and sensitizes glioblastoma cells to tumor necrosis factor (TNFα)-induced apoptosis through SIRT1 inhibition. Cell Death Dis. 2012;3:e271.
- Pallares J, Llobet D, Santacana M, et al. CK2beta is expressed in endometrial carcinoma and has a role in apoptosis resistance and cell proliferation. Am J Pathol. 2009;174(1):287–296.
- Prudent R, Moucadel V, Nguyen C-H, et al. Antitumor activity of pyridocarbazole and benzopyridoindole derivatives that inhibit protein kinase CK2. Cancer Res. 2010;70(23):9865–9874.
- Wang G, Ahmad KA, Ahmed K. Modulation of death receptor-mediated apoptosis by CK2. Mol Cell Biochem. 2005;274(1–2):201–205.
- Ruzzene M, Di Maira G, Tosoni K, et al. Assessment of CK2 constitutive activity in cancer cells. Methods Enzymol. 2010;484:495–514.
- Di Maira G, Brustolon F, Tosoni K, et al. Comparative analysis of CK2 expression and function in tumor cell lines displaying sensitivity vs. resistance to chemical induced apoptosis. Mol Cell Biochem. 2008;316(1–2):155–161.
- Kreutzer JN, Ruzzene M, Guerra B. Enhancing chemosensitivity to gemcitabine via RNA interference targeting the catalytic subunits of protein kinase CK2 in human pancreatic cancer cells. BMC Cancer. 2010;10:440.
- Piazza FA, Ruzzene M, Gurrieri C, et al. Multiple myeloma cell survival relies on high activity of protein kinase CK2. Blood. 2006;108(5):1698–1707.
- Siddiqui-Jain A, Drygin D, Streiner N, et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010;70(24):10288–10298.
- Buontempo F, McCubrey JA, Orsini E, et al. Therapeutic targeting of CK2 in acute and chronic leukemias. Leukemia. 2018;32(1):1–10.
- Silva A, Jotta PY, Silveira AB, et al. Regulation of PTEN by CK2 and Notch1 in primary T-cell acute lymphoblastic leukemia: rationale for combined use of CK2- and gamma-secretase inhibitors. Haematologica. 2010;95(4):674–678.
- Silva A, Yunes JA, Cardoso BA, et al. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J Clin Invest. 2008;118(11):3762–3774.
- Kim JS, Eom JI, Cheong J-W, et al. Protein kinase CK2alpha as an unfavorable prognostic marker and novel therapeutic target in acute myeloid leukemia. Clin Cancer Res. 2007;13(3):1019–1028.
- Martins LR, Lúcio P, Silva MC, et al. Targeting CK2 overexpression and hyperactivation as a novel therapeutic tool in chronic lymphocytic leukemia. Blood. 2010;116(15):2724–2731.
- Karin M, Delhase M. The I kappa B kinase (IKK) and NF-kappa B: key elements of proinflammatory signalling. Semin Immunol. 2000;12(1):85–98.
- McElhinny JA, Trushin SA, Bren GD, et al. Casein kinase II phosphorylates I kappa B alpha at S-283, S-289, S-293, and T-291 and is required for its degradation. Mol Cell Biol. 1996;16(3):899–906.
- Schwarz KB. Oxidative stress during viral infection: a review. Free Radic Biol Med. 1996;21(5):641–649.
- Shen Y, Kapfhamer D, Minnella AM, et al. Bioenergetic state regulates innate inflammatory responses through the transcriptional co-repressor CtBP. Nat Commun. 2017;8(1):624.
- Baldwin AS. Jr., The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683.
- Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441(7092):431–436.
- Perkins ND, Gilmore TD. Good cop, bad cop: the different faces of NF-kappaB. Cell Death Differ. 2006;13(5):759–772.
- Quotti Tubi L, Canovas Nunes S, Brancalion A, et al. Protein kinase CK2 regulates AKT, NF-κB and STAT3 activation, stem cell viability and proliferation in acute myeloid leukemia. Leukemia. 2017;31(2):292–300.
- Bird TA, Schooley K, Dower SK, et al. Activation of nuclear transcription factor NF-kappaB by interleukin-1 is accompanied by casein kinase II-mediated phosphorylation of the p65 subunit. J Biol Chem. 1997;272(51):32606–32612.
- Wang D, Westerheide SD, Hanson JL, et al. Tumor necrosis factor alpha-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J Biol Chem. 2000;275(42):32592–32597.
- Chantome A, Pance A, Gauthier N, et al. Casein kinase II-mediated phosphorylation of NF-kappaB p65 subunit enhances inducible nitric-oxide synthase gene transcription in vivo. J Biol Chem. 2004;279(23):23953–23960.
- Parhar K, Ray A, Steinbrecher U, et al. The p38 mitogen-activated protein kinase regulates interleukin-1beta-induced IL-8 expression via an effect on the IL-8 promoter in intestinal epithelial cells. Immunology. 2003;108(4):502–512.
- Hagemann T, Lawrence T, McNeish I, et al. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med. 2008;205(6):1261–1268.
- Nakagawa MM, Chen H, Rathinam CV. Constitutive activation of NF-κB pathway in hematopoietic stem cells causes loss of quiescence and deregulated transcription factor networks. Front Cell Dev Biol. 2018;6:143.
- Connelly L, Barham W, Onishko HM, et al. NF-kappaB activation within macrophages leads to an anti-tumor phenotype in a mammary tumor lung metastasis model. Breast Cancer Res. 2011;13(4):R83.
- Pagliari LJ, Perlman H, Liu H, et al. Macrophages require constitutive NF-kappaB activation to maintain A1 expression and mitochondrial homeostasis. Mol Cell Biol. 2000;20(23):8855–8865.
- Feuerer M, Hill JA, Mathis D, et al. Foxp3+ regulatory T cells: differentiation, specification, subphenotypes. Nat Immunol. 2009;10(7):689–695.
- Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2(10):725–734.
- Liu T, Zhang L, Joo D, et al. NF-kappaB signaling in inflammation. Signal Transduct Target Ther. 2017;2:17023.
- Groner B, von Manstein V. Jak Stat signaling and cancer: opportunities, benefits and side effects of targeted inhibition. Mol Cell Endocrinol. 2017;451:1–14.
- Hedvat M, Huszar D, Herrmann A, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16(6):487–497.
- Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282(28):20059–20063.
- Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809.
- Zheng Y, Qin H, Frank SJ, et al. A CK2-dependent mechanism for activation of the JAK-STAT signaling pathway. Blood. 2011;118(1):156–166.
- Carter-Su C, Argetsinger LS. JAKs, Stats, and CK2? Blood. 2011;118(1):5–6.
- Yang J, Liao X, Agarwal MK, et al. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 2007;21(11):1396–1408.
- Kaplan MH, Grusby MJ. Regulation of T helper cell differentiation by STAT molecules. J Leukoc Biol. 1998;64(1):2–5.
- Pernis AB, Rothman PB. JAK-STAT signaling in asthma. J Clin Invest. 2002;109(10):1279–1283.
- Lio CW, Hsieh CS. A two-step process for thymic regulatory T cell development. Immunity. 2008;28(1):100–111.
- Yang XO, Panopoulos AD, Nurieva R, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282(13):9358–9363.
- Avery DT, Ma CS, Bryant VL, et al. STAT3 is required for IL-21-induced secretion of IgE from human naive B cells. Blood. 2008;112(5):1784–1793.
- Rivas-Caicedo A, Soldevila G, Fortoul TI, et al. Jak3 is involved in dendritic cell maturation and CCR7-dependent migration. PLoS One. 2009;4(9):e7066.
- Pattison MJ, Mackenzie KF, Arthur JS. Inhibition of JAKs in macrophages increases lipopolysaccharide-induced cytokine production by blocking IL-10-mediated feedback. J Immunol. 2012;189(6):2784–2792.
- Hodge DR, Xiao W, Wang LH, et al. Activating mutations in STAT3 and STAT5 differentially affect cellular proliferation and apoptotic resistance in multiple myeloma cells. Cancer Biol Ther. 2004;3(2):188–194.
- Hubert A, Paris S, Piret J-P, et al. Casein kinase 2 inhibition decreases hypoxia-inducible factor-1 activity under hypoxia through elevated p53 protein level. J Cell Sci. 2006;119(Pt 16):3351–3362.
- Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9(6):685–693.
- Mottet D, Ruys SPD, Demazy C, et al. Role for casein kinase 2 in the regulation of HIF-1 activity. Int J Cancer. 2005;117(5):764–774.
- Pluemsampant S, Safronova OS, Nakahama K-i, et al. Protein kinase CK2 is a key activator of histone deacetylase in hypoxia-associated tumors. Int J Cancer. 2008;122(2):333–341.
- Jung YJin, Isaacs JS, Lee S, et al. Hypoxia-inducible factor induction by tumour necrosis factor in normoxic cells requires receptor-interacting protein-dependent nuclear factor kappa B activation. Biochem J. 2003;370(Pt 3):1011–1017.
- Lu X-G, Xing C-G, Feng Y-Z, et al. Clinical significance of immunohistochemical expression of hypoxia-inducible factor-1alpha as a prognostic marker in rectal adenocarcinoma. Clin Colorectal Cancer. 2006;5(5):350–353.
- Wenger RH. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. Faseb J. 2002;16(10):1151–1162.
- Kong T, Eltzschig HK, Karhausen J, et al. Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of beta2 integrin gene expression. Proc Natl Acad Sci USA. 2004;101(28):10440–10445.
- Silva-Pavez E, Tapia JC. Protein kinase CK2 in cancer energetics. Front Oncol. 2020;10:893.
- Mirzoeva S, Kim ND, Chiu K, et al. Inhibition of HIF-1 alpha and VEGF expression by the chemopreventive bioflavonoid apigenin is accompanied by Akt inhibition in human prostate carcinoma PC3-M cells. Mol Carcinog. 2008;47(9):686–700.
- Cramer T, Johnson RS. A novel role for the hypoxia inducible transcription factor HIF-1alpha: critical regulation of inflammatory cell function. Cell Cycle. 2003;2(3):192–193.
- Karhausen J, Furuta GT, Tomaszewski JE, et al. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest. 2004;114(8):1098–1106.
- Jantsch J, Chakravortty D, Turza N, et al. Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J Immunol. 2008;180(7):4697–4705.
- Kojima H, Sitkovsky MV, Cascalho M. HIF-1 alpha deficiency perturbs T and B cell functions. Curr Pharm Des. 2003;9(23):1827–1832.
- Lukashev D, Klebanov B, Kojima H, et al. Cutting edge: hypoxia-inducible factor 1alpha and its activation-inducible short isoform I.1 negatively regulate functions of CD4+ and CD8+ T lymphocytes. J Immunol. 2006;177(8):4962–4965.
- Nelson N, Szekeres K, Iclozan C, et al. Apigenin: selective CK2 inhibitor increases Ikaros expression and improves T cell homeostasis and function in murine pancreatic cancer. PLoS One. 2017;12(2):e0170197.
- Bakhle YS. COX-2 and cancer: a new approach to an old problem. Br J Pharmacol. 2001;134(6):1137–1150.
- Hashemi Goradel N, Najafi M, Salehi E, et al. Cyclooxygenase-2 in cancer: a review. J Cell Physiol. 2019;234(5):5683–5699.
- Yefi R, Ponce DP, Niechi I, et al. Protein kinase CK2 promotes cancer cell viability via up-regulation of cyclooxygenase-2 expression and enhanced prostaglandin E2 production. J Cell Biochem. 2011;112(11):3167–3175.
- Khan MS, Halagowder D, Devaraj SN. Methylated chrysin induces co-ordinated attenuation of the canonical Wnt and NF-kB signaling pathway and upregulates apoptotic gene expression in the early hepatocarcinogenesis rat model. Chem Biol Interact. 2011;193(1):12–21.
- Liu B, Qu L, Yan S. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int. 2015;15:106.
- Telliez A, Furman C, Pommery N, et al. Mechanisms leading to COX-2 expression and COX-2 induced tumorigenesis: topical therapeutic strategies targeting COX-2 expression and activity. Anticancer Agents Med Chem. 2006;6(3):187–208.
- Kambayashi T, et al. Potential involvement of IL-10 in suppressing tumor-associated macrophages. Colon-26-derived prostaglandin E2 inhibits TNF-alpha release via a mechanism involving IL-10. J Immunol. 1995;154(7):3383–3390.
- Stolina M, Sharma S, Lin Y, et al. Specific inhibition of cyclooxygenase 2 restores antitumor reactivity by altering the balance of IL-10 and IL-12 synthesis. J Immunol. 2000;164(1):361–370.
- Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182(8):4499–4506.
- Serafini P. Editorial: PGE2-producing MDSC: a role in tumor progression? J Leukoc Biol. 2010;88(5):827–829.
- Chou JP, Ramirez CM, Ryba DM, et al. Prostaglandin E2 promotes features of replicative senescence in chronically activated human CD8+ T cells. PLoS One. 2014;9(6):e99432.
- Yang L, Yamagata N, Yadav R, et al. Cancer-associated immunodeficiency and dendritic cell abnormalities mediated by the prostaglandin EP2 receptor. J Clin Invest. 2003;111(5):727–735.
- Avruch J. MAP kinase pathways: the first twenty years. Biochim Biophys Acta. 2007;1773(8):1150–1160.
- Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9(7):517–531.
- Sharma S, Stolina M, Yang S-C, et al. Tumor cyclooxygenase 2-dependent suppression of dendritic cell function. Clin Cancer Res. 2003;9(3):961–968.
- Khotskaya YB, Holla VR, Farago AF, et al. Targeting TRK family proteins in cancer. Pharmacol Ther. 2017;173:58–66.
- Plotnikov A, Chuderland D, Karamansha Y, et al. Nuclear extracellular signal-regulated kinase 1 and 2 translocation is mediated by casein kinase 2 and accelerated by autophosphorylation. Mol Cell Biol. 2011;31(17):3515–3530.
- Rubinfeld H, Seger R. The ERK cascade: a prototype of MAPK signaling. Mol Biotechnol. 2005;31(2):151–174.
- Bang YJ, Kwon JH, Kang SH, et al. Increased MAPK activity and MKP-1 overexpression in human gastric adenocarcinoma. Biochem Biophys Res Commun. 1998;250(1):43–47.
- Bhartiya D, Singh J. FSH-FSHR3-stem cells in ovary surface epithelium: basis for adult ovarian biology, failure, aging, and cancer. Reproduction. 2015;149(1):R35–R48.
- Tang Q, Wu J, Zheng F, et al. Emodin increases expression of insulin-like growth factor binding protein 1 through activation of MEK/ERK/AMPKα and interaction of PPARγ and Sp1 in lung cancer. Cell Physiol Biochem. 2017;41(1):339–357.
- Dumitru CD, Ceci JD, Tsatsanis C, et al. TNF-alpha induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway. Cell. 2000;103(7):1071–1083.
- Jorritsma PJ, Brogdon JL, Bottomly K. Role of TCR-induced extracellular signal-regulated kinase activation in the regulation of early IL-4 expression in naive CD4+ T cells. J Immunol. 2003;170(5):2427–2434.
- Yamashita M, Kimura M, Kubo M, et al. T cell antigen receptor-mediated activation of the Ras/mitogen-activated protein kinase pathway controls interleukin 4 receptor function and type-2 helper T cell differentiation. Proc Natl Acad Sci USA. 1999;96(3):1024–1029.
- Qian C, Qian L, Yu Y, et al. Fas signal promotes the immunosuppressive function of regulatory dendritic cells via the ERK/β-catenin pathway. J Biol Chem. 2013;288(39):27825–27835.
- Yasuda T, Kometani K, Takahashi N, et al. ERKs induce expression of the transcriptional repressor Blimp-1 and subsequent plasma cell differentiation. Sci Signal. 2011;4(169):ra25.
- Andersson ER, Sandberg R, Lendahl U. Notch signaling: simplicity in design, versatility in function. Development. 2011;138(17):3593–3612.
- Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. 2011;11(5):338–351.
- Zhang S, Long H, Yang Y-L, et al. Inhibition of CK2α down-regulates Notch1 signalling in lung cancer cells. J Cell Mol Med. 2013;17(7):854–862.
- Kulbe H, Iorio F, Chakravarty P, et al. Integrated transcriptomic and proteomic analysis identifies protein kinase CK2 as a key signaling node in an inflammatory cytokine network in ovarian cancer cells. Oncotarget. 2016;7(13):15648–15661.
- Zhang S, Yang Y, Liang Z, et al. Silybin-mediated inhibition of Notch signaling exerts antitumor activity in human hepatocellular carcinoma cells. PLoS One. 2013;8(12):e83699.
- Parker KH, Beury DW, Ostrand-Rosenberg S. Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv Cancer Res. 2015;128:95–139.
- Cheng P, Kumar V, Liu H, et al. Effects of notch signaling on regulation of myeloid cell differentiation in cancer. Cancer Res. 2014;74(1):141–152.
- Chiplunkar SV, Gogoi D. The multifaceted role of Notch signal in regulating T cell fate. Immunol Lett. 2019;206:59–64.
- Gibb DR, Saleem SJ, Kang D-J, et al. ADAM10 overexpression shifts lympho- and myelopoiesis by dysregulating site 2/site 3 cleavage products of Notch. J Immunol. 2011;186(7):4244–4252.
- Sauma D, Ramirez A, Alvarez K, et al. Notch signalling regulates cytokine production by CD8+ and CD4+ T cells. Scand J Immunol. 2012;75(4):389–400.
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–1274.
- Cheung M, Testa JR. Diverse mechanisms of AKT pathway activation in human malignancy. Curr Cancer Drug Targets. 2013;13(3):234–244.
- Di Maira G, Salvi M, Arrigoni G, et al. Protein kinase CK2 phosphorylates and upregulates Akt/PKB. Cell Death Differ. 2005;12(6):668–677.
- Guerra B. Protein kinase CK2 subunits are positive regulators of AKT kinase. Int J Oncol. 2006;28(3):685–693.
- Ruzzene M, Bertacchini J, Toker A, et al. Cross-talk between the CK2 and AKT signaling pathways in cancer. Adv Biol Regul. 2017;64:1–8.
- Samuels Y, Waldman T. Oncogenic mutations of PIK3CA in human cancers. Curr Top Microbiol Immunol. 2010;347:21–41.
- Covarrubias AJ, Aksoylar HI, Horng T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin Immunol. 2015;27(4):286–296.
- Koyasu S. The role of PI3K in immune cells. Nat Immunol. 2003;4(4):313–319.
- Okkenhaug K, Bilancio A, Farjot G, et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science. 2002;297(5583):1031–1034.
- Xie J, Qian J, Yang J, et al. Critical roles of Raf/MEK/ERK and PI3K/AKT signaling and inactivation of p38 MAP kinase in the differentiation and survival of monocyte-derived immature dendritic cells. Exp Hematol. 2005;33(5):564–572.
- Lee YG, Lee J, Byeon SE, et al. Functional role of Akt in macrophage-mediated innate immunity. Front Biosci. 2011;16:517–530.
- Abu-Eid R, Samara RN, Ozbun L, et al. Selective inhibition of regulatory T cells by targeting the PI3K-Akt pathway. Cancer Immunol Res. 2014;2(11):1080–1089.
- Kim EH, Sullivan JA, Plisch EH, et al. Signal integration by Akt regulates CD8 T cell effector and memory differentiation. J Immunol. 2012;188(9):4305–4314.
- Nagai S, Kurebayashi Y, Koyasu S. Role of PI3K/Akt and mTOR complexes in Th17 cell differentiation. Ann N Y Acad Sci. 2013;1280:30–34.
- Pilon-Thomas S, Nelson N, Vohra N, et al. Murine pancreatic adenocarcinoma dampens SHIP-1 expression and alters MDSC homeostasis and function. PLoS One. 2011;6(11):e27729.
- Georgopoulos K, Bigby M, Wang JH, et al. The Ikaros gene is required for the development of all lymphoid lineages. Cell. 1994;79(1):143–156.
- Merkenschlager M. Ikaros in immune receptor signaling, lymphocyte differentiation, and function. FEBS Lett. 2010;584(24):4910–4914.
- Gurel Z, Ronni T, Ho S, et al. Recruitment of ikaros to pericentromeric heterochromatin is regulated by phosphorylation. J Biol Chem. 2008;283(13):8291–8300.
- Popescu M, Gurel Z, Ronni T, et al. Ikaros stability and pericentromeric localization are regulated by protein phosphatase 1. J Biol Chem. 2009;284(20):13869–13880.
- Wang H, Ouyang H, Lai L, et al. Pathogenesis and regulation of cellular proliferation in acute lymphoblastic leukemia – the role of Ikaros. J Buon. 2014;19(1):22–28.
- Song C, Gowda C, Pan X, et al. Targeting casein kinase II restores Ikaros tumor suppressor activity and demonstrates therapeutic efficacy in high-risk leukemia. Blood. 2015;126(15):1813–1822.
- Cho S-J, Huh J-E, Song J, et al. Ikaros negatively regulates inducible nitric oxide synthase expression in macrophages: involvement of Ikaros phosphorylation by casein kinase 2. Cell Mol Life Sci. 2008;65(20):3290–3303.
- Dominguez I, Sonenshein GE, Seldin DC. Protein kinase CK2 in health and disease: CK2 and its role in Wnt and NF-kappaB signaling: linking development and cancer. Cell Mol Life Sci. 2009;66(11–12):1850–1857.
- de Groot REA, Rappel SB, Lorenowicz MJ, et al. Protein kinase CK2 is required for Wntless internalization and Wnt secretion. Cell Signal. 2014;26(12):2601–2605.
- Seldin DC, Landesman-Bollag E, Farago M, et al. CK2 as a positive regulator of Wnt signalling and tumourigenesis. Mol Cell Biochem. 2005;274(1–2):63–67.
- Song DH, Sussman DJ, Seldin DC. Endogenous protein kinase CK2 participates in Wnt signaling in mammary epithelial cells. J Biol Chem. 2000;275(31):23790–23797.
- Wang B, Tian T, Kalland K-H, et al. Targeting Wnt/β-catenin signaling for cancer immunotherapy. Trends Pharmacol Sci. 2018;39(7):648–658.
- Luke JJ, Bao R, Sweis RF, et al. WNT/β-catenin pathway activation correlates with immune exclusion across human cancers. Clin Cancer Res. 2019;25(10):3074–3083.
- Swafford D, Manicassamy S. Wnt signaling in dendritic cells: its role in regulation of immunity and tolerance. Discov Med. 2015;19(105):303–310.
- Hong Y, Manoharan I, Suryawanshi A, et al. Deletion of LRP5 and LRP6 in dendritic cells enhances antitumor immunity. Oncoimmunology. 2016;5(4):e1115941.
- Gattinoni L, Zhong X-S, Palmer DC, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009;15(7):808–813.
- Sun X, Liu S, Wang D, et al. Colorectal cancer cells suppress CD4+ T cells immunity through canonical Wnt signaling. Oncotarget. 2017;8(9):15168–15181.
- Yde CW, Frogne T, Lykkesfeldt AE, et al. Induction of cell death in antiestrogen resistant human breast cancer cells by the protein kinase CK2 inhibitor DMAT. Cancer Lett. 2007;256(2):229–237.
- Pagano MA, Bain J, Kazimierczuk Z, et al. The selectivity of inhibitors of protein kinase CK2: an update. Biochem J. 2008;415(3):353–365.
- Meggio F, Pagano MA, Moro S, et al. Inhibition of protein kinase CK2 by condensed polyphenolic derivatives. An in vitro and in vivo study. Biochemistry. 2004;43(40):12931–12936.
- Zandomeni R, Zandomeni MC, Shugar D, et al. Casein kinase type II is involved in the inhibition by 5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole of specific RNA polymerase II transcription. J Biol Chem. 1986;261(7):3414–3419.
- Sarno S, Ruzzene M, Frascella P, et al. Development and exploitation of CK2 inhibitors. Mol Cell Biochem. 2005;274(1–2):69–76.
- Yim H, Lee YH, Lee CH, et al. Emodin, an anthraquinone derivative isolated from the rhizomes of Rheum palmatum, selectively inhibits the activity of casein kinase II as a competitive inhibitor. Planta Med. 1999;65(1):9–13.
- Meggio F, Shugar D, Pinna LA. Ribofuranosyl-benzimidazole derivatives as inhibitors of casein kinase-2 and casein kinase-1. Eur J Biochem. 1990;187(1):89–94.
- Sarno S, Reddy H, Meggio F, et al. Selectivity of 4,5,6,7-tetrabromobenzotriazole, an ATP site-directed inhibitor of protein kinase CK2 (‘casein kinase-2’). FEBS Lett. 2001;496(1):44–48.
- Lian H, Su M, Zhu Y, et al. Protein kinase CK2, a potential therapeutic target in carcinoma management. Asian Pac J Cancer Prev. 2019;20(1):23–32.
- Quotti Tubi L, Gurrieri C, Brancalion A, et al. Inhibition of protein kinase CK2 with the clinical-grade small ATP-competitive compound CX-4945 or by RNA interference unveils its role in acute myeloid leukemia cell survival, p53-dependent apoptosis and daunorubicin-induced cytotoxicity. J Hematol Oncol. 2013;6:78.
- Perea SE, Baladrón I, Valenzuela C, et al. CIGB-300: a peptide-based drug that impairs the Protein Kinase CK2-mediated phosphorylation. Semin Oncol. 2018;45(1–2):58–67.
- Benavent Acero F, Capobianco CS, Garona J, et al. CIGB-300, an anti-CK2 peptide, inhibits angiogenesis, tumor cell invasion and metastasis in lung cancer models. Lung Cancer. 2017;107:14–21.
- Martins LR, Perera Y, Lúcio P, et al. Targeting chronic lymphocytic leukemia using CIGB-300, a clinical-stage CK2-specific cell-permeable peptide inhibitor. Oncotarget. 2014;5(1):258–263.
- Hashimoto A, Gao C, Mastio J, et al. Inhibition of casein kinase 2 disrupts differentiation of myeloid cells in cancer and enhances the efficacy of immunotherapy in mice. Cancer Res. 2018;78(19):5644–5655.
- Jung M, Park KH, Kim HM, et al. Inhibiting casein kinase 2 overcomes paclitaxel resistance in gastric cancer. Gastric Cancer. 2019;22(6):1153–1163.
- Zhou B, Ritt DA, Morrison DK, et al. Protein kinase CK2α maintains extracellular signal-regulated kinase (ERK) activity in a CK2α kinase-independent manner to promote resistance to inhibitors of RAF and MEK but Not ERK in BRAF mutant melanoma. J Biol Chem. 2016;291(34):17804–17815.
- Parker R, Clifton-Bligh R, Molloy MP. Phosphoproteomics of MAPK inhibition in BRAF-mutated cells and a role for the lethal synergism of dual BRAF and CK2 inhibition. Mol Cancer Ther. 2014;13(7):1894–1906.
- Bandyopadhyay K, Gjerset RA. Protein kinase CK2 is a central regulator of topoisomerase I hyperphosphorylation and camptothecin sensitivity in cancer cell lines. Biochemistry. 2011;50(5):704–714.
- Bandyopadhyay K, Li P, Gjerset RA. CK2-mediated hyperphosphorylation of topoisomerase I targets serine 506, enhances topoisomerase I-DNA binding, and increases cellular camptothecin sensitivity. PLoS One. 2012;7(11):e50427.
- Dubois N, Willems M, Nguyen-Khac M-T, et al. Constitutive activation of casein kinase 2 in glioblastomas: absence of class restriction and broad therapeutic potential. Int J Oncol. 2016;48(6):2445–2452.
- Intemann J, Saidu NEB, Schwind L, et al. ER stress signaling in ARPE-19 cells after inhibition of protein kinase CK2 by CX-4945. Cell Signal. 2014;26(7):1567–1575.