
Abstract
Non-alcoholic fatty liver disease/steatohepatitis (NAFLD/NASH) are becoming major liver diseases worldwide. Liver fibrosis and cirrhosis are among the most significant risk factors of hepatocellular carcinoma (HCC) and associated with the long-term prognosis of NAFLD patients. To stratify the risk of HCC in NAFLD patients clinically, the discovery of non-invasive fibrosis markers is needed urgently. Liver macrophages play critical roles in the regulation of inflammation and fibrosis by interacting with hepatic stellate cells (HSCs) and other immune cells. Thus, it is rational to explore feasible biomarkers for liver fibrosis by focusing on macrophage-related factors. We examined serum factors comprehensively in multiple cohorts of NAFLD/NASH patients to determine whether they were correlated with the biopsy-proven fibrosis stage. We found that the serum levels of interleukin (IL)-34, YKL-40 and soluble Siglec-7 (sSiglec7) were closely associated with liver fibrosis and served as diagnostic biomarkers in patients with NAFLD/NASH. In the NAFLD liver, IL-34 was produced by activated fibroblasts, and YKL-40 and sSiglec-7 were secreted from macrophages. The sensitivity and specificity of these markers to detect advanced liver fibrosis varied, supporting the notion that the combination of these markers with other modalities is an option for clinical application.
Keywords:
1. Introduction
Hepatocellular carcinoma (HCC) is the sixth most commonly diagnosed cancer and the fourth leading cause of cancer-related deaths worldwide [Citation1]. The burden of HCC is highest in Asia and Africa, and its incidence and mortality have been increasing dramatically in Western countries. In 2015, the reported number of new cases of liver cancer was 854,000 and that of liver cancer-related deaths was 810,000 [Citation1]. Reduction of HCC-related deaths and prevention of the progression from chronic hepatitis to liver cirrhosis are among the major targets for management in various countries. Hepatitis viruses, such as hepatitis B virus (HBV) and HCV, have been considered as the main causes of liver cancer. Recently, the proportion of HCC caused by non-viral etiologies, including alcohol and steatohepatitis, has been increasing worldwide [Citation1,Citation2]. A study on the prevalence of HCC in waitlisted candidates for liver transplantation showed that non-alcoholic fatty liver disease (NAFLD)-related HCC patients had increased by 11.5-fold from 2002 to 2016, while that of HCV-related HCC patients had increased by 6.2-fold [Citation3]. It has been reported that concomitant metabolic syndrome increases the risk of liver cirrhosis and HCC in patients with chronic HBV infection [Citation4,Citation5]. Currently, HCV can be eradicated in more than 95% of patients by direct anti-viral agents [Citation6]. Even in patients who have been cleared of HCV, the risk of hepato-carcinogenesis continues to be high in those with moderate/advanced liver fibrosis or metabolic disorders, such as steatosis and diabetes [Citation7,Citation8]. Therefore, regardless of etiology, correction of metabolic derangement should be a priority to reduce the risk of HCC.
Development of liver fibrosis is a continuous phenomenon accompanied by inflammation and tissue remodeling characterized by excessive accumulation of extracellular matrix (ECM), which can eventually lead to liver cirrhosis. Hepatic stellate cells (HSCs) are major players in ECM deposition and the dominant source of myofibroblasts [Citation9]. Upon liver injury of any etiology, quiescent HSCs undergo a phenotypic switch, known as activation, leading to acquisition of a myofibroblast-like phenotype. In this process, macrophages play crucial roles in the activation of HSCs [Citation10]. Liver cirrhosis is the primary risk factor for HCC independent of liver disease etiology [Citation11–13]. It is estimated that one-third of cirrhotic patients eventually develop liver cancer [Citation14]. Therefore, defining the patient population with a high risk of HCC, such as patients with cirrhosis or advanced fibrosis, is of clinical importance to screen early-stage HCC. In this review, we describe several basic and clinical studies including ours on macrophage-related biomarkers of liver fibrosis and HCC in patients with NAFLD and discuss their potential effects on clinical practice.
2. Increasing trend of NAFLD-related HCC
NAFLD is recognized as the most common cause of chronic liver disease, the proportion of which in the general population is estimated to be 15–30% worldwide [Citation15]. NAFLD is a metabolic syndrome often complicated by cardiovascular disease, obesity, diabetes and dyslipidemia. Unless the metabolic dysfunction is corrected, NAFLD is deemed as a progressive disease similar to chronic viral hepatitis, shifting from simple steatosis, steatohepatitis and liver cirrhosis to HCC. Non-alcoholic steatohepatitis (NASH), which is found in 10–20% of NAFLD patients, is considered as a serious form of NAFLD. A definitive diagnosis of NASH is based on histological evidence of not only fat accumulation (steatosis) in hepatocytes, but also liver cell injury and death as well as accumulation of inflammatory cells [Citation16]. The risk of developing end-stage liver cirrhosis and HCC is increased compared with simple steatosis. The fibrosis progression rate (FPR) in patients with NAFLD or NASH was estimated by a meta-analysis [Citation17]. The overall FPRs, or stage 1 progression determined by a paired biopsy, were 14.3 years for NAFLD and 7.1 years for NASH [Citation17]. Although patients with NASH appear to have a lower risk of HCC than those with HCV-related cirrhosis, the annual incidence is approximately 1–2%. In a large cohort study on Veterans with NASH cirrhosis in the United States, the incidence of HCC was 1.06 per 100 person-years [Citation1]. Of particular importance, studies have shown that one-fourth of NASH-related HCC occurs in the absence of cirrhosis [Citation18,Citation19]. However, other studies have shown that the annual incidence rate of HCC in non-cirrhotic NASH is low. A study on the Veterans Affairs health system revealed a HCC incidence of only 0.008 person-years in a cohort of 292,366 patients with non-cirrhotic NAFLD [Citation13]. Similarly, data from Taiwan showed that the 5-year cumulative incidence of HCC from non-cirrhosis was 1.0% [Citation20]. Such discordant reports may be influenced by the heterologous definition of NAFLD, different proportions of patients with metabolic syndrome, different genetic predisposition due to ethnicity and observational bias.
A nationwide survey for the etiology of liver cirrhosis in Japan revealed that non-viral cirrhosis (non-B, non-C; NBNC), including alcohol- and NASH-related cirrhosis, had notably increased from 2008 to 2016. Such proportional changes of alcohol- and NASH-related cirrhosis increased from 13.7 to 24.9% and 2.0 to 9.1%, respectively, in Japan [Citation21]. Similar to this trend, the proportion of liver cancer with non-viral etiology has been increasing significantly in Japan. According to a cumulative study on the etiology of HCC in Japan, the proportion of NBNC liver cancer had increased to 32.5% in 2015, 15% of which was related to NAFLD [Citation2]. In a 2010 national survey of HCC in Japan, the development rate of NASH-HCC from cirrhosis was 11.3%/5 years, which was lower than that of HCV-HCC (30.5%/5 years) [Citation22].
3. NAFLD/NASH is a multifaceted and progressive liver disease
Based on altered lipid metabolism and excessive lipid accumulation in hepatocytes, multiple causative factors are involved in the pathogenesis of NAFLD, such as genetic susceptibility variants, obesity, insulin resistance, type 2 DM (T2DM), hyperlipidemia, hypertension and changes in gut microbiota ().
Figure 1. Multifaceted mechanisms in the progression from steatosis to fibrosis/cirrhosis/HCC in non-alcoholic fatty liver disease. The major players involved in the pathogenesis of NAFLD/NASH are shown. The details are described in the text. CAF: cancer-associated fibroblasts; DAMPs: danger-associated molecular patterns; HCC: hepatocellular carcinoma; NAFLD: non-alcoholic fatty liver disease; NASH: non-alcoholic steatohepatitis; PAMPs: pathogen-associated molecular patterns; TAM: tumor-associated macrophage.
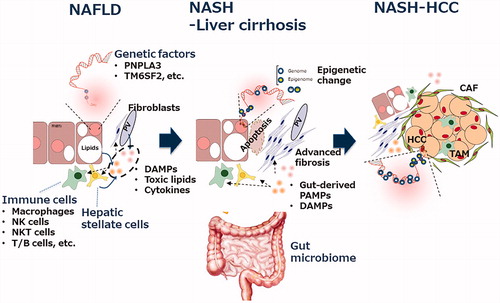
3.1. Genetic factors
NAFLD/NASH has complex disease traits such that interactions between the environment and susceptible polygenic host factors determine the disease phenotype and influence progression [Citation23]. Pathological drivers are unlikely to be identical among all patients. One of the most investigated genetic variants associated with the progression of NAFLD is PNPLA3. PNPLA3 is a lipid-metabolizing enzyme and its variant, I148M, is associated with fat accumulation, inflammation and susceptibility to steatohepatitis. From its functional viewpoint, it has been reported that PNPLA3 is required for HSC activation and the I148M variant potentiates the profibrogenic phenotype by enhancing CCL5 and GM-CSF expression [Citation24]. These data suggest a beneficial effect of downregulating the PNPLA3 148 M variant in individuals at risk of NASH. The other genes reported so far are TM6SF2, MBOAT7 and GCKR, which are involved in lipid metabolism or insulin resistance [Citation25]. Epigenetic regulation can be caused by alterations in DNA methylation, modifications to histone proteins, chromatin remodeling and non-coding RNAs, the involvement of which in the pathogenesis of NAFLD/NASH is reviewed elsewhere [Citation25].
3.2. Metabolic factors
NAFLD/NASH is a liver disease associated with metabolic syndrome [Citation23]. The prevalence of NAFLD is proportional to an increase in body mass index (BMI). In fact, the prevalence of NAFLD in the general population is about 25%, but increases to over 90% for very obese (BMI ≥35 kg/m2) individuals. Compared with the NAFLD population in Western countries, there are more lean NAFLD patients in Japan and other Asian countries, comprising 15–27% of non-obese populations [Citation26]. Several clinical, metabolic, histological and gut microbiome features are reported to be different between classical and lean NAFLD patients, suggesting that metabolic adaptation is distinct between the groups [Citation27]. It is yet to be determined whether the clinical outcomes are different, including the development of cirrhosis and HCC, between obese and lean NAFLD. To define pathological drivers of NAFLD/NASH based on the concept of lipotoxicity, comprehensive analysis of lipid species in the NAFLD liver is underway [Citation28]. Luukkonen et al. reported that, by comparing the hepatic lipidome in NAFLD patients, the saturated and ceramide-enriched lipid profile was associated with an increased risk of T2DM and cardiovascular disease [Citation29].
3.3. Microbiome
A significant effect of the microbiome on the pathogenesis of NAFLD/NASH has been reported by several studies of humans and mice [Citation30]. Gut microbiota affects hepatic carbohydrates, lipid metabolism and the balance between proinflammatory and anti-inflammatory effects. After transferring the fecal microbiome from diseased mice with fasting hyperglycemia and insulinemia to germ-free healthy mice, NAFLD developed in recipient mice [Citation31]. Bacterial species, such as Escherichia and Bacteroides, are higher in patients with NASH compared with matched healthy individuals [Citation32,Citation33]. An increase in Proteobacteria and a decrease in Firmicutes are observed during progression of NAFLD, suggesting that the gut microbiome is not constantly dysbiotic [Citation34]. A dysfunctional gut barrier and altered gut permeability are observed in NAFLD patients [Citation34–36]. Recently, Ni et al. reported that nuclear receptor Rev-erbα is involved in the regulation of intestinal permeability in NASH [Citation37]. Induction of a leaky and inflamed gut by dextran sulfate sodium leads to translocation of lipopolysaccharide (LPS) to systemic circulation and consequently worsens liver inflammation and fibrosis in mice fed a high-fat diet [Citation38]. Similarly, JAM-A-deficient mice, a genetic model of gut barrier dysfunction, fed a high fat, fructose and cholesterol diet develop more severe steatohepatitis than control mice [Citation39]. Gut microbiota promotes weight gain and hepatic steatosis through Farnesoid X receptor-dependent mechanisms [Citation40].
3.4. Cytokines and chemokines
Serum levels of tumor necrosis factor-alpha (TNF-α), CCL2/MCP-1, interleukin-1β (IL-1β) and IL-6 are elevated in NAFLD patients compared with healthy controls [Citation41–43]. Such higher levels of TNF-α and CCL2/MCP-1 are observed in patients with NASH than in those with simple steatosis [Citation43]. TNF-α is overexpressed in the liver and adipose tissue of NASH patients [Citation42]. The promotor variant regulating TNF-α expression is enriched in Italian patients with NAFLD [Citation44]. IL-15, which activate NK cells, has been reported to be elevated in NAFLD patients [Citation45,Citation46] and IL-15 is implicated in the high-fat diet-induced lipid accumulation and inflammation in NASH [Citation47]. In mouse models and patients with NAFLD, CXCL10 is upregulated and involved in steatohepatitis and hepatocyte injury, which is mediated by proinflammatory cytokines and the NF-κB pathway [Citation48]. In sera and biopsy specimens from NAFLD patients, CXCL16 is upregulated [Citation49]. In vitro, inhibition of CXCL16 reduces infiltration of inflammatory macrophages into the liver and decreases intrahepatic levels of TNF-α and CCL2/MCP-1 [Citation50].
3.5. Macrophages
Increased numbers of macrophages are observed in the liver of NAFLD patients with severe fibrosis [Citation38,Citation39]. An increase of portal macrophages is the earliest change at the steatosis stage before inflammation or fibrosis and predicts disease progression in patients with NAFLD [Citation40]. These reports raise the possibility that the abundance of activated macrophages in the liver is accompanied by the progression of NAFLD. Therefore, macrophage-related biomarkers may be feasible to diagnose liver inflammation and fibrosis or stratification of patients according to the risk of fibrosis progression.
Liver macrophages include resident Kupffer cells and monocyte-derived cells. In mice, Kupffer cells are defined as F4/80+CD11bmid and monocyte-derived macrophages are defined as F4/80midCD11b+ [Citation51]. In a human NAFLD study, CCR2 was highly expressed in monocyte-derived macrophages, and CCR2+ macrophages were increased in patients with NAFLD [Citation52,Citation53]. During NAFLD, gut-derived endotoxins (e.g., LPS), gut-derived pathogen-associated molecular patterns (PAMPs), fatty acids, cholesterol and its metabolites, as well as damage-associated molecular patterns (DAMPs) are released from damaged hepatocytes and activate Kupffer cells that in turn secrete inflammatory cytokines (e.g., TNF-α and IL-1β) and chemokines (e.g., CCL2) [Citation54,Citation55]. CCL2 and other chemokines promote infiltration of CCR2+Ly-6C+ monocytes into the damaged liver. Experimental mouse models of NASH have shown that infiltration of Ly-6C+ monocytes is a critical pathological event promoting steatohepatitis and subsequent fibrosis progression in NASH [Citation52,Citation56,Citation57]. During chronic injury, Ly-6C+ monocytes mainly produce TGF-β that activates HSCs to become collagen-producing myofibroblasts [Citation58]. In the crosstalk between macrophages and HSCs, it has been reported that macrophage c-mer tyrosine kinase (Mer-TK) receptor promotes an ERK-TGF-β1 pathway that activates HSCs and liver fibrosis [Citation59]. A genetic study has shown that a variant of the Mer-TK gene is associated with lower hepatic expression of Mer-TK and a reduced risk of fibrosis progression in patients with NASH [Citation60].
3.6. NK cells
NK cells are abundant in the liver and play pivotal roles in the regulation of inflammation and fibrosis. The functions of NK cells are controlled by cytokines and their multiple activation (e.g., NKG2D and NKp46) or inhibitory (e.g., NKG2A and KIR) receptors [Citation61]. Activated NK cells are reported to exert an inhibitory effect against liver fibrosis by killing activated HSCs or decreasing infiltration of monocyte-derived macrophages into the liver [Citation62–64]. Crosstalk between macrophages and NK cells has been implicated in the pathogenesis of NASH. In a mouse model of NAFLD, NKp46+ NK cells potentially prevented fibrosis by regulating M1/M2 polarization of liver macrophages [Citation65].
3.7. T/B cells
The cellular role of adaptive immunity, such as T and B cells, in the pathogenesis of NAFLD/NASH has yet to be clarified. The involvement of oxidative stress in driving NAFLD-associated immune responses was identified by elevated titers of anti-oxidative stress-derived epitope IgG in ∼40% of adult patients with NAFLD [Citation66,Citation67]. Chronic inflammation and fibrosis in humans and mice with NAFLD are accompanied by accumulation of liver-resident immunoglobulin-A-producing B (IgA+ B) cells. IgA+ B cells are capable of suppressing liver cytotoxic CD8+ T cells, which may be associated with prevention of HCC [Citation68].
4. Assessment of liver fibrosis in NAFLD/NASH patients
Histological staging of liver fibrosis is the most influential predictor of long-term mortality and liver-related events in NAFLD patients [Citation69]. Therefore, evaluating fibrosis is crucial to manage patients with NAFLD/NASH, especially those at risk of HCC. Liver biopsy remains as the gold standard to evaluate the degree of hepatic necro-inflammation and fibrosis in patients with chronic liver disease. However, such an invasive procedure has substantial limitations. Fatal complications are sometimes unavoidable, such as bleeding or biliary tract infection. Furthermore, repetitive biopsies for sequential assessment of a disease may not be possible in clinical practice. Thus, several non-invasive investigations have been developed to establish the diagnosis of liver inflammation or fibrosis and evaluate the treatment response [Citation70].
In NAFLD patients, platelet count is a simple indicator for advanced fibrosis [Citation71]. Furthermore, extensive studies have been conducted on other indicators, such as the fibrosis score [fibrosis-4 (FIB-4], NAFLD fibrosis score (NFS), AST-to-platelet ratio index, gamma-glutamyl transpeptidase-to-platelet ratio, serum biomarkers (type-IV collagen, hyaluronic acid and Mac-2-binding protein glycosylation isomer, ultrasound-based elastography (virtual touch tissue quantification and transient elastography) and magnetic resonance elastography (MRE) [Citation72–76]. FIB-4 has been regarded as a useful score to identify advanced fibrosis in NAFLD patients. Clinical algorithms have been proposed to diagnose and follow patients with NAFLD using a combination of fibrosis indicators [Citation72,Citation77]. In the algorithm, NAFLD patients are divided into three groups (low/intermediate/high risk) using FIB-4 or NFS. For patients in the intermediate group, ultrasound-based elastography or MRE are recommended, and liver biopsy should be considered for those in the high-risk group [Citation77].
5. Exploration of macrophage-related biomarkers for liver fibrosis
In the machineries of fibrogenesis in NASH, activation of macrophages and HSCs as well as and their interactions might play essential roles as described in the previous section. Therefore, it is hypothesized that biological factors related to these cells may be indicators of macrophage and HSC activation, and serve as fibrosis-related markers in patients with NAFLD/NASH.
5.1. sCD163
Scavenger receptor CD163 (sCD163), the IL-6 inducible macrophage receptor for clearing haptoglobin–hemoglobin complexes, is expressed on macrophages and upregulated by macrophage activation [Citation78]. The soluble form (sCD163) is shed from activated macrophages and released into the bloodstream [Citation79]. Increased levels of serum sCD163 are observed in patients with various diseases, such as sepsis, myeloid leukemia, tuberculosis and HIV infection [Citation80–82]. Soluble CD163 levels may serve as a predictor of disease severity and clinical outcomes of fulminant hepatic failure and alcoholic hepatitis [Citation83,Citation84]. Soluble CD163 levels are increased in association with the fibrosis stage in patients with chronic HBV or HCV infection and NAFLD [Citation85–87]. In NAFLD patients, hepatic expression of CD163 correlates with the degree of steatosis [Citation88]. Soluble CD163 parallels plasma free fatty acid levels and lipolysis in adipose tissue [Citation88], suggesting crosstalk between adipose tissue and liver macrophages in NAFLD.
5.2. IL-34
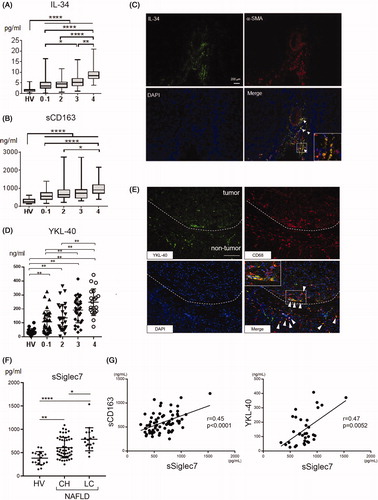
IL-34 is a ligand for colony-stimulating factor-1 receptor (CSF-1R). IL-34 binds to CSF-1R and promotes differentiation, proliferation and survival of monocytes and macrophages, the function of which is the same as the other CSF-1R ligand, macrophage colony-stimulating factor (M-CSF) [Citation89]. During HCV infection, Preisser et al. showed that serum levels of IL-34 and M-CSF are higher in patients with F3-F4 (advanced fibrosis) than in those with F0–F2, suggesting that these cytokines are profibrotic factors [Citation90]. We comprehensively evaluated serum levels of macrophage-related markers (IL-34, M-CSF and soluble CD163) and 40 cytokines/chemokines in 197 liver biopsy-proven NAFLD patients with various fibrosis stages [Citation91]. IL-34 was increased significantly with the progression of fibrosis and was an independent marker of liver cirrhosis in addition to sCD163 (). Serum IL-34 levels were positively correlated with sCD163 [Citation91]. To diagnose liver cirrhosis, the area under the curve, sensitivity and specificity of IL-34 (0.87, 83.3% and 80.2%, respectively) were superior to or comparable with other serum biomarkers and fibrosis indexes. Positive correlations were observed between IL-34 and other fibrosis markers, such as FIB-4 and the NAFLD activity score. Immunostaining revealed that almost all IL-34 positive cells in the liver tissue were fibroblasts (α-SMA-positive cells) (). Thus, we demonstrated that IL-34 is feasible to evaluate the degree of fibrosis in NAFLD patients. Noda et al. reported that IL-34 is a useful biomarker for not only fibrosis, but also risk stratification of the prognosis in patients with non-viral HCC [Citation92]. They showed that HCC patients with high IL-34 levels had poorer prognoses than those with low IL-34 levels.
Figure 2. Associations of the serum levels of sCD163, IL-34, YKL40 and sSiglec7 with liver fibrosis in NAFLD patients. A, B) Serum levels of IL-34 (A) and sCD163 (B) in healthy volunteers (n = 20) and biopsy-proven NAFLD patients (n = 197). Horizontal axis: 0–1, 2, 3 and 4 depict the stages of liver fibrosis (F0–1, F2, F3 and F4, respectively). *p≤ .05; **p≤.01; ****p≤.0001 by the Kruskal–Wallis test with Dunn’s multiple comparison test. HV: healthy volunteer. C) Liver tissue was obtained from patients with NAFLD (liver cirrhosis/stage 4). Immunofluorescence staining of frozen liver tissues from patients with NAFLD is shown (×200, green, IL-34; red, α -SMA; blue, DAPI). White arrowheads mark the positions of IL-34- and α-SMA-positive cells. Inset in the photomicrograph shows fluorescent merged cells at an enlarged scale. D) Serum levels of YKL-40 in healthy volunteers (n = 19) and biopsy-proven NAFLD patients (n = 111). Horizontal axis: 0–1, 2, 3 and 4 depict the stages of liver fibrosis (F0–1, F2, F3 and F4, respectively). **p≤ .01 by the Kruskal–Wallis test with Dunn’s multiple comparison test. HV: healthy volunteer. E) Immunofluorescence staining of YKL-40 and CD68 in frozen liver specimens from NAFLD patients with HCC. The dotted line indicates the margin between tumor and non-tumor tissues. White arrowheads mark the positions of YKL-40- and CD68-positive cells. The inset of the photomicrograph shows fluorescent merged cells at an enlarged scale. Scale bar represents 100 μm (×200, green, YKL-40; red, CD68; blue, DAPI). F) Serum levels of sSiglec7 in healthy volunteers (N = 19) and NAFLD patients (n = 93) with chronic hepatitis and liver cirrhosis. Mean ± SD. Horizontal axis: 0–1, 2, 3 and 4 depict the stages of liver fibrosis (F0–1, F2, F3 and F4, respectively). *p<.05; **p<.01; ****p<.0001 by the Kruskal–Wallis test with Dunn’s multiple comparison test. HV: healthy volunteer; CH: chronic hepatitis; LC: liver cirrhosis. G) Correlation of serum sSiglec7 with sCD163 and YKL-40 in NAFLD/NASH patients. Correlations were assessed by Spearman’s analysis.
5.3. YKL-40
YKL-40 (also known as chitinase 3-like-1, breast regression protein-39 and human cartilage glycoprotein-39) is a chitinase-like protein that is found in humans as a secretion product of articular chondrocytes and synovial cells [Citation93]. Serum YKL-40 levels are elevated in patients with a wide variety of human diseases characterized by sustained inflammation, including rheumatoid arthritis, atherosclerosis, type 2 diabetes, chronic pancreatitis, asthma and Crohn’s disease [Citation94–97]. YKL-40 is expressed by various cell types including neutrophils [Citation98], macrophages [Citation99] and several types of malignant tumors [Citation100]. The serum YKL-40 level has been evaluated as a noninvasive marker of various chronic inflammatory and fibrotic liver diseases including chronic hepatitis C [Citation101], chronic hepatitis B [Citation102] and alcoholic liver disease [Citation103]. Because YKL-40 is secreted by activated macrophages, it is believed to act as a chemoattractant for endothelial cells and modulate angiogenesis during tissue repair, which is also expressed in multiple tissues including the human liver [Citation104–106]. We measured serum YKL-40 levels in 111 NAFLD patients and 23 HCC patients with NAFLD [Citation91]. Serum YKL-40 levels in NAFLD patients were increased in accordance with the progression of liver fibrosis (). Multivariate analysis revealed that YKL-40 was an independent factor that was significantly associated with severe fibrosis (F3–4). Serum YKL-40 levels in HCC patients with non-cirrhotic NAFLD were significantly higher than in those without HCC. Immunofluorescence staining showed that YKL-40 was expressed by macrophages in liver tissue of NAFLD patients. We also found that peritumoral macrophages expressed YKL-40 in liver tissue from NAFLD patients with HCC () [Citation91].
5.4. Soluble Siglec-7 (sSiglec7)
Sialic acid-binding immunoglobulin-like lectins (Siglecs) are a family of cell surface proteins expressed mainly on innate immune cells including macrophages and NK cells [Citation107,Citation108]. Siglecs are molecules that recognize a characteristic profile of sialic acids and regulate cellular activity by transmitting inhibitory or activation signals [Citation109]. Macrophages express Siglec-1, -3, -8, -9, -11, -15 and -16 depending on their differentiation status [Citation110–112]. However, in the settings of liver disease, the expression profile of Siglecs on macrophages is still largely unknown. It has been reported that Siglec-7-positive NK cells are decreased in patients with HBV, HCV or HIV infections [Citation113,Citation114]. In addition, serum soluble Siglec-7 (sSiglec-7) was increased in such patients and positively correlated with liver fibrosis in HBV- and HCV-infected patients [Citation113]. Based on these results, it has been speculated that sSiglec-7 is shed from Siglec-7+ NK cells activated by viruses [Citation113,Citation114].
Serum sSiglec-7 was significantly increased in NAFLD patients compared with healthy volunteers, the levels of which were correlated positively with sCD163 and YKL-40 (). Serum sSiglec-7 was an independent diagnostic marker and had an initial divergence variable with high specificity (95.4%) for advanced fibrosis (F3 and F4) in NAFLD patients. In livers from NAFLD patients, inflammatory CCR2+ macrophages increase in parallel to the NASH severity and fibrosis stage [Citation52]. We showed that liver CCR2+ macrophages highly express Siglec-7, while Tim4+ resident macrophages express Siglec-7 weakly [Citation53]. Therefore, serum sSiglec-7, reflecting macrophage activation, can serve as an independent marker of advanced liver fibrosis with high specificity in patients with NAFLD. In terms of the function of the soluble form, it has been reported that sSiglec-7 is capable of binding HIV-1 and enhances the susceptibility of CD4+ T cells and monocyte-derived macrophages to infection [Citation114]. The effect of sSiglec-7 on the pathogenesis of chronic liver diseases, including NAFLD and viral hepatitis, needs to be explored further.
6. Characteristics of macrophage-related biomarkers for liver fibrosis
We showed that serum levels of sCD163, YKL-40, IL-34 and sSiglec-7 could be feasible for diagnostic biomarkers for liver fibrosis in patients with NAFLD/NASH [Citation53,Citation91]. Of interest, sCD163, YKL-40 and sSiglec-7 are mainly produced by inflammatory macrophages, and IL-34 is produced from activated fibroblasts, respectively. The important issues that need to be addressed are: 1) What are the comparative diagnostic performance of each marker and 2) Are these better than conventional fibrosis index like FIB-4.
sSiglec-7, not sCD163 and YKL-40, was an independent predictor for advanced fibrosis (F3–4). Decision-tree algorithm using age, platelet counts, sSiglec-7, sCD163, YKL-40 and FIB-4 index for advanced fibrosis showed that sSiglec-7 was the first divergent variable. Age was the second classifier, and YKL-40 was the third classifier [Citation53]. These results suggested that sSiglec-7 is more specific for the diagnosis of advanced fibrosis compared to sCD163, YKL-40 or FIB-4.
IL-34 was superior to sCD163 and FIB-4 index as a diagnostic marker of fibrosis in patients with NAFLD [Citation91]. Serum YKL-40 levels were higher in NASH-LC patients with HCC than those in NASH-LC patients without HCC [Citation115]. Therefore, sSiglec-7, IL-34 and YKL-40 might be useful for diagnosing advanced fibrosis, mild/moderate fibrosis and HCC in NAFLD patients, respectively.
7. Perspectives
Advanced fibrosis, cirrhosis and HCC are the major liver-related clinical endpoints of patients with NAFLD/NASH. It is well noted that prevention of fibrosis progression should be the target for management of NAFLD patients. Evidence-based practice guidelines published by international and domestic associations for liver disease have recommended that dietary changes and lifestyle intervention should be the first-line therapies for such patients [Citation116–118]. Extensive basic and clinical studies have provided useful information on the mechanisms of liver inflammation, fibrosis progression and carcinogenesis. Of particular importance, macrophages arguably play crucial but dichotomous roles in the pathogenesis of NAFLD by promoting fibrogenesis, while enhancing fibrinolysis in the resolution phase. Such a distinct capacity of macrophages may be influenced by their cellular origin, metabolic factors and the liver microenvironment, and tightly regulated by distinct sets of transcription factors. Therefore, macrophages have been gaining much attention as not only a source of diagnostic biomarkers, as described here, but also a therapeutic target against liver disease [Citation10]. A novel macrophage-oriented drug in the NASH pipeline is Cenicriviroc (CCR2/5 dual antagonist), the pharmacological effect of which is inhibition of the infiltration and accumulation of bone marrow-derived macrophages in the liver. A phase 2b study on Cenicriviroc appears promising, showing an anti-fibrotic effect in some patients with advanced fibrosis caused by NASH [Citation119]. Further profound understanding of the heterogenous and complex nature of macrophages may lead to establishment of novel diagnostic and therapeutic strategies against hard-to-treat NASH cirrhosis and liver cancer.
Acknowledgments
The authors thank Mitchell Arico from Edanz Group (https://en-author-services.edanzgroup.com/) for editing a draft of this manuscript.
Disclosure statement
Tatsuya Kanto has received lecture fees from Gilead Sciences and MSD.
Additional information
Funding
References
- Singal AG, Lampertico P, Nahon P. Epidemiology and surveillance for hepatocellular carcinoma: nNew trends. J Hepatol. 2020;72(2):250–261.
- Tateishi R, Uchino K, Fujiwara N, et al. A nationwide survey on non-B, non-C hepatocellular carcinoma in Japan: 2011–2015 update. J Gastroenterol. 2019;54(4):367–376.
- Younossi ZM, Stepanova M, Ong J, et al.; Global NASH Council. Effects of alcohol consumption and metabolic syndrome on mortality in patients with nonalcoholic and alcohol-related fatty liver disease. Clin Gastroenterol Hepatol. 2019;17(8):1625–1633.e1.
- Wong GL-H, Wong VW-S, Choi PC-L, et al. Metabolic syndrome increases the risk of liver cirrhosis in chronic hepatitis B. Gut. 2009;58(1):111–117.
- Kim NH, Cho YK, Kim BI, et al. Effect of metabolic syndrome on the clinical outcomes of chronic hepatitis B patients with nucleos(t)ide analogues treatment. Dig Dis Sci. 2018;63(10):2792–2799.
- Webster DP, Klenerman P, Dusheiko GM, et al. Hepatitis C. Lancet. 2015;385(9973):1124–1135.
- Hung C-H, Lee C-M, Wang J-H, et al. Impact of diabetes mellitus on incidence of hepatocellular carcinoma in chronic hepatitis C patients treated with interferon-based antiviral therapy. Int J Cancer. 2011;128(10):2344–2352.
- Peleg N, Issachar A, Sneh Arbib O, et al. Liver steatosis is a major predictor of poor outcomes in chronic hepatitis C patients with sustained virological response. J Viral Hepat. 2019;26(11):1257–1265.
- Schwabe RF, Tabas I, Pajvani UB. Mechanisms of fibrosis development in NASH. Gastroenterology. 2020;158(7):1913–1928.
- Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. 2017;66(6):1300–1312.
- Kanwal F, Kramer J, Asch SM, et al. Risk of hepatocellular cancer in HCV patients treated with direct-acting antiviral agents. Gastroenterology. 2017;153(4):996–1005.e1.
- Fattovich G, Stroffolini T, Zagni I, et al. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology. 2004;127(5 Suppl 1):S35–S50.
- Kanwal F, Kramer JR, Mapakshi S, et al. Risk of hepatocellular cancer in patients with non-alcoholic fatty liver disease. Gastroenterology. 2018;155(6):1828–1837.e2.
- Sangiovanni A, Prati GM, Fasani P, et al. The natural history of compensated cirrhosis due to hepatitis C virus: a 17-year cohort study of 214 patients. Hepatology. 2006;43(6):1303–1310.
- Bellentani S. The epidemiology of non-alcoholic fatty liver disease. Liver Int. 2017;37(Suppl 1):81–84.
- Diehl AM, Day C. Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. N Engl J Med. 2017;377(21):2063–2072.
- Singh S, Allen AM, Wang Z, et al. Fibrosis progression in nonalcoholic fatty liver vs. nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin Gastroenterol Hepatol. 2015;13(4):643–654.e1–9; quiz e39-40.
- Mittal S, El-Serag HB, Sada YH, et al. Hepatocellular carcinoma in the absence of cirrhosis in United States veterans is associated with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2016;14(1):124–131.e1.
- Dyson J, Jaques B, Chattopadyhay D, et al. Hepatocellular cancer: the impact of obesity, type 2 diabetes and a multidisciplinary team. J Hepatol. 2014;60(1):110–117.
- Lee TY, Wu JC, Yu SH, et al. The occurrence of hepatocellular carcinoma in different risk stratifications of clinically noncirrhotic nonalcoholic fatty liver disease. Int J Cancer. 2017;141(7):1307–1314.
- Enomoto H, Ueno Y, Hiasa Y, et al.; Japan Etiology of Liver Cirrhosis Study Group in the 54th Annual Meeting of JSH. Transition in the etiology of liver cirrhosis in Japan: a nationwide survey. J Gastroenterol. 2020;55(3):353–362.
- Tokushige K, Hashimoto E, Kodama K. Hepatocarcinogenesis in non-alcoholic fatty liver disease in Japan. J Gastroenterol Hepatol. 2013;28(Suppl 4):88–92.
- Cotter TG, Rinella M. NAFLD 2020: the state of the disease. Gastroenterology. 2020;158(7):1851–1864.
- Bruschi FV, Claudel T, Tardelli M, et al. The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells. Hepatology. 2017;65(6):1875–1890.
- Eslam M, Valenti L, Romeo S. Genetics and epigenetics of NAFLD and NASH: clinical impact. J Hepatol. 2018;68(2):268–279.
- Seto WK, Yuen MF. Nonalcoholic fatty liver disease in Asia: emerging perspectives. J Gastroenterol. 2017;52(2):164–174.
- Chen F, Esmaili S, Rogers GB, et al. Lean NAFLD: a distinct entity shaped by differential metabolic adaptation. Hepatology. 2019;71(4):1213–1227.
- Musso G, Cassader M, Paschetta E, et al. Bioactive lipid species and metabolic pathways in progression and resolution of nonalcoholic steatohepatitis. Gastroenterology. 2018;155(2):282–302.e8.
- Luukkonen PK, Zhou Y, Sädevirta S, et al. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J Hepatol. 2016;64(5):1167–1175.
- Kolodziejczyk AA, Zheng D, Shibolet O, et al. The role of the microbiome in NAFLD and NASH. EMBO Mol Med. 2019;11(2):e9302.
- Le Roy T, Llopis M, Lepage P, et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut. 2013;62(12):1787–1794.
- Zhu L, Baker SS, Gill C, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. 2013;57(2):601–609.
- Boursier J, Mueller O, Barret M, et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology. 2016;63(3):764–775.
- Loomba R, Seguritan V, Li W, et al. Gut microbiome-based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab. 2017;25(5):1054–1062.e5.
- Miele L, Valenza V, La Torre G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49(6):1877–1887.
- Volynets V, Küper MA, Strahl S, et al. Nutrition, intestinal permeability, and blood ethanol levels are altered in patients with nonalcoholic fatty liver disease (NAFLD). Dig Dis Sci. 2012;57(7):1932–1941.
- Ni Y, Zhao Y, Ma L, et al. Pharmacological activation of REV-ERBα improves nonalcoholic steatohepatitis by regulating intestinal permeability. Metabolism. 2021;114:154409.
- Gabele E, et al. DSS induced colitis increases portal LPS levels and enhances hepatic inflammation and fibrogenesis in experimental NASH. J Hepatol. 2011;55(6):1391–1399.
- Rahman K, Desai C, Iyer SS, et al. Loss of junctional adhesion molecule a promotes severe steatohepatitis in mice on a diet high in saturated fat, fructose, and cholesterol. Gastroenterology. 2016;151(4):733–746.e12.
- Parseus A, et al. Microbiota-induced obesity requires farnesoid X receptor. Gut. 2017;66(3):429–437.
- Bertola A, Bonnafous S, Anty R, et al. Hepatic expression patterns of inflammatory and immune response genes associated with obesity and NASH in morbidly obese patients. PLoS One. 2010;5(10):e13577.
- Crespo J. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology. 2001;34(6):1158–1163.
- Haukeland JW, Damås JK, Konopski Z, et al. Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J Hepatol. 2006;44(6):1167–1174.
- Valenti L, Fracanzani AL, Dongiovanni P, et al. Tumor necrosis factor alpha promoter polymorphisms and insulin resistance in nonalcoholic fatty liver disease. Gastroenterology. 2002;122(2):274–280.
- Carson WE, Giri JG, Lindemann MJ, et al. Interleukin (IL) 15 is a novel cytokine that activates human natural killer cells via components of the IL-2 receptor. J Exp Med. 1994;180(4):1395–1403.
- Wada N, Takaki A, Ikeda F, et al. Serum-inducible protein (IP)-10 is a disease progression-related marker for non-alcoholic fatty liver disease. Hepatol Int. 2017;11(1):115–124.
- Cepero-Donates Y, Lacraz G, Ghobadi F, et al. Interleukin-15-mediated inflammation promotes non-alcoholic fatty liver disease. Cytokine. 2016;82:102–111.
- Zhang X, Shen J, Man K, et al. CXCL10 plays a key role as an inflammatory mediator and a non-invasive biomarker of non-alcoholic steatohepatitis. J Hepatol. 2014;61(6):1365–1375.
- Jiang L, Yang M, Li X, et al. CXC motif ligand 16 promotes nonalcoholic fatty liver disease progression via hepatocyte-stellate cell crosstalk. J Clin Endocrinol Metab. 2018;103(11):3974–3985.
- Wehr A, Baeck C, Ulmer F, et al. Pharmacological inhibition of the chemokine CXCL16 diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. PLoS One. 2014;9(11):e112327.
- Matsuda M, Tsurusaki S, Miyata N, et al. Oncostatin M causes liver fibrosis by regulating cooperation between hepatic stellate cells and macrophages in mice. Hepatology. 2018;67(1):296–312.
- Krenkel O, Puengel T, Govaere O, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology. 2018;67(4):1270–1283.
- Sakamoto Y, Yoshio S, Doi H, et al. Serum soluble sialic acid-binding immunoglobulin-like lectin-7 concentration as an indicator of liver macrophage activation and advanced fibrosis in patients with non-alcoholic fatty liver disease. Hepatol Res. 2020;50(4):466–477.
- Kazankov K, Jørgensen SMD, Thomsen KL, et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol. 2019;16(3):145–159.
- Tosello-Trampont A-C, Landes SG, Nguyen V, et al. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-α production. J Biol Chem. 2012;287(48):40161–40172.
- Baeck C, Wehr A, Karlmark KR, et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut. 2012;61(3):416–426.
- Reid DT, Reyes JL, McDonald BA, et al. Kupffer cells undergo fundamental changes during the development of experimental NASH and are critical in initiating liver damage and inflammation. PLoS One. 2016;11(7):e0159524.
- Karlmark KR, Weiskirchen R, Zimmermann HW, et al. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology. 2009;50(1):261–274.
- Cai B, Dongiovanni P, Corey KE, et al. Macrophage MerTK promotes liver fibrosis in nonalcoholic steatohepatitis. Cell Metab. 2020;31(2):406–421.e7.
- Petta S, Valenti L, Marra F, et al. MERTK rs4374383 polymorphism affects the severity of fibrosis in non-alcoholic fatty liver disease. J Hepatol. 2016;64(3):682–690.
- Liu P, Chen L, Zhang H. Natural killer cells in liver disease and hepatocellular carcinoma and the NK cell-based immunotherapy. J Immunol Res. 2018;2018:1206737.
- Radaeva S, Sun R, Jaruga B, et al. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology. 2006;130(2):435–452.
- Fan Y, Zhang W, Wei H, et al. Hepatic NK cells attenuate fibrosis progression of non-alcoholic steatohepatitis in dependent of CXCL10-mediated recruitment. Liver Int. 2020;40(3):598–608.
- Wijaya RS, Read SA, Schibeci S, et al. KLRG1+ natural killer cells exert a novel antifibrotic function in chronic hepatitis B. J Hepatol. 2019;71(2):252–264.
- Tosello-Trampont A-C, Krueger P, Narayanan S, et al. NKp46(+) natural killer cells attenuate metabolism-induced hepatic fibrosis by regulating macrophage activation in mice. Hepatology. 2016;63(3):799–812.
- Sutti S, Jindal A, Locatelli I, et al. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology. 2014;59(3):886–897.
- Albano E. Immune response towards lipid peroxidation products as a predictor of progression of non-alcoholic fatty liver disease to advanced fibrosis. Gut. 2005;54(7):987–993.
- Shalapour S, Lin X-J, Bastian IN, et al. Inflammation-induced IgA + cells dismantle anti-liver cancer immunity. Nature. 2017;551(7680):340–345.
- Angulo P, Kleiner DE, Dam-Larsen S, et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology. 2015;149(2):389–397.e10.
- Vilar-Gomez E, Calzadilla-Bertot L, Wai-Sun Wong V, et al. Fibrosis severity as a determinant of cause-specific mortality in patients with advanced nonalcoholic fatty liver disease: a multi-national cohort study. Gastroenterology. 2018;155(2):443–457.e17.
- Yoneda M, Fujii H, Sumida Y, et al.; Japan Study Group of Nonalcoholic Fatty Liver Disease. Platelet count for predicting fibrosis in nonalcoholic fatty liver disease. J Gastroenterol. 2011;46(11):1300–1306.
- Yoneda M, Imajo K, Takahashi H, et al. Clinical strategy of diagnosing and following patients with nonalcoholic fatty liver disease based on invasive and noninvasive methods. J Gastroenterol. 2018;53(2):181–196.
- Li Q, Lu C, Li W, et al. The gamma-glutamyl transpeptidase to platelet ratio for non-invasive assessment of liver fibrosis in patients with chronic hepatitis B and non-alcoholic fatty liver disease. Oncotarget. 2017;8(17):28641–28649.
- Abe M, Miyake T, Kuno A, et al. Association between Wisteria floribunda agglutinin-positive Mac-2 binding protein and the fibrosis stage of non-alcoholic fatty liver disease. J Gastroenterol. 2015;50(7):776–784.
- Yoneda M, Yoneda M, Yoneda M, et al. Transient elastography in patients with non-alcoholic fatty liver disease (NAFLD). Gut. 2007;56(9):1330–1331.
- Dulai PS, Sirlin CB, Loomba R. MRI and MRE for non-invasive quantitative assessment of hepatic steatosis and fibrosis in NAFLD and NASH: Clinical trials to clinical practice. J Hepatol. 2016;65(5):1006–1016.
- Vilar-Gomez E, Chalasani N. Non-invasive assessment of non-alcoholic fatty liver disease: clinical prediction rules and blood-based biomarkers. J Hepatol. 2018;68(2):305–315.
- Moestrup SK, Moller HJ. CD163: a regulated hemoglobin scavenger receptor with a role in the anti-inflammatory response. Ann Med. 2004;36(5):347–354.
- Weaver LK, Hintz-Goldstein KA, Pioli PA, et al. Pivotal advance: activation of cell surface Toll-like receptors causes shedding of the hemoglobin scavenger receptor CD163. J Leukoc Biol. 2006;80(1):26–35.
- Moller HJ, Hald K, Moestrup SK. Characterization of an enzyme-linked immunosorbent assay for soluble CD163. Scand J Clin Lab Invest. 2002;62(4):293–299.
- Knudsen TB, Gustafson P, Kronborg G, et al. Predictive value of soluble haemoglobin scavenger receptor CD163 serum levels for survival in verified tuberculosis patients. Clin Microbiol Infect. 2005;11(9):730–735.
- Burdo TH, Lentz MR, Autissier P, et al. Soluble CD163 made by monocyte/macrophages is a novel marker of HIV activity in early and chronic infection prior to and after anti-retroviral therapy. J Infect Dis. 2011;204(1):154–163.
- Hiraoka A, Horiike N, Akbar SMF, et al. Soluble CD163 in patients with liver diseases: very high levels of soluble CD163 in patients with fulminant hepatic failure. J Gastroenterol. 2005;40(1):52–56.
- Saha B, Tornai D, Kodys K, et al. Biomarkers of macrophage activation and immune danger signals predict clinical outcomes in alcoholic hepatitis. Hepatology. 2019;70(4):1134–1149.
- Kazankov K, Barrera F, Møller HJ, et al. Soluble CD163, a macrophage activation marker, is independently associated with fibrosis in patients with chronic viral hepatitis B and C. Hepatology. 2014;60(2):521–530.
- Kazankov K, Tordjman J, Møller HJ, et al. Macrophage activation marker soluble CD163 and non-alcoholic fatty liver disease in morbidly obese patients undergoing bariatric surgery. J Gastroenterol Hepatol. 2015;30(8):1293–1300.
- Kazankov K, Barrera F, Møller HJ, et al. The macrophage activation marker sCD163 is associated with morphological disease stages in patients with non-alcoholic fatty liver disease. Liver Int. 2016;36(10):1549–1557.
- Rosso C, Kazankov K, Younes R, et al. Crosstalk between adipose tissue insulin resistance and liver macrophages in non-alcoholic fatty liver disease. J Hepatol. 2019;71(5):1012–1021.
- Lin H, Lee E, Hestir K, et al. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science. 2008;320(5877):807–811.
- Preisser L, Miot C, Le Guillou-Guillemette H, et al. IL-34 and macrophage colony-stimulating factor are overexpressed in hepatitis C virus fibrosis and induce profibrotic macrophages that promote collagen synthesis by hepatic stellate cells. Hepatology. 2014;60(6):1879–1890.
- Shoji H, Yoshio S, Mano Y, et al. Interleukin-34 as a fibroblast-derived marker of liver fibrosis in patients with non-alcoholic fatty liver disease. Sci Rep. 2016;6:28814.
- Noda Y, Kawaguchi T, Korenaga M, et al. High serum interleukin-34 level is a predictor of poor prognosis in patients with non-viral hepatocellular carcinoma. Hepatol Res. 2019;49(9):1046–1053.
- Hakala BE, White C, Recklies AD. Human cartilage gp-39, a major secretory product of articular chondrocytes and synovial cells, is a mammalian member of a chitinase protein family. J Biol Chem. 1993;268(34):25803–25810.
- Johansen JS. Serum YKL-40 concentrations in patients with rheumatoid arthritis: relation to disease activity. Rheumatology (Oxford). 1999;38(7):618–626.
- Rathcke CN, Johansen JS, Vestergaard H. YKL-40, a biomarker of inflammation, is elevated in patients with type 2 diabetes and is related to insulin resistance. Inflamm Res. 2006;55(2):53–59.
- Konradsen JR, James A, Nordlund B, et al. The chitinase-like protein YKL-40: a possible biomarker of inflammation and airway remodeling in severe pediatric asthma. J Allergy Clin Immunol. 2013;132(2):328–335.e5.
- Erzin Y, Uzun H, Karatas A, et al. Serum YKL-40 as a marker of disease activity and stricture formation in patients with Crohn’s disease. J Gastroenterol Hepatol. 2008;23(8 Pt 2):e357–e362.
- Volck B, Price PA, Johansen JS, et al. YKL-40, a mammalian member of the chitinase family, is a matrix protein of specific granules in human neutrophils. Proc Assoc Am Physicians. 1998;110(4):351–360.
- Rehli M, Krause SW, Andreesen R. Molecular characterization of the gene for human cartilage gp-39 (CHI3L1), a member of the chitinase protein family and marker for late stages of macrophage differentiation. Genomics. 1997;43(2):221–225.
- Johansen JS, Jensen BV, Roslind A, et al. Serum YKL-40, a new prognostic biomarker in cancer patients? Cancer Epidemiol Biomarkers Prev. 2006;15(2):194–202.
- Kamal SM, Turner B, He Q, et al. Progression of fibrosis in hepatitis C with and without schistosomiasis: correlation with serum markers of fibrosis. Hepatology. 2006;43(4):771–779.
- Lee KG, Seo YS, An H, et al. Usefulness of non-invasive markers for predicting liver cirrhosis in patients with chronic hepatitis B. J Gastroenterol Hepatol. 2010;25(1):94–100.
- Nojgaard C, et al. Serum levels of YKL-40 and PIIINP as prognostic markers in patients with alcoholic liver disease. J Hepatol. 2003;39(2):179–186.
- Sztrolovics R, Recklies AD, Roughley PJ, et al. Hyaluronate degradation as an alternative mechanism for proteoglycan release from cartilage during interleukin-1beta-stimulated catabolism. Biochem J. 2002;362(Pt 2):473–479.
- Malinda KM, Ponce L, Kleinman HK, et al. Gp38k, a protein synthesized by vascular smooth muscle cells, stimulates directional migration of human umbilical vein endothelial cells. Exp Cell Res. 1999;250(1):168–173.
- Lee CG, Da Silva CA, Dela Cruz CS, et al. Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annu Rev Physiol. 2011;73:479–501.
- Duan S, Paulson JC. Siglecs as immune cell checkpoints in disease. Annu Rev Immunol. 2020;38:365–395.
- Crocker PR, Clark EA, Filbin M, et al. Siglecs: a family of sialic-acid binding lectins. Glycobiology. 1998;8(2):v.
- Crocker PR, McMillan SJ, Richards HE. CD33-related siglecs as potential modulators of inflammatory responses. Ann NY Acad Sci. 2012;1253:102–111.
- Cao H, Lakner U, de Bono B, et al. SIGLEC16 encodes a DAP12-associated receptor expressed in macrophages that evolved from its inhibitory counterpart SIGLEC11 and has functional and non-functional alleles in humans. Eur J Immunol. 2008;38(8):2303–2315.
- Morinaga H, Mayoral R, Heinrichsdorff J, et al. Characterization of distinct subpopulations of hepatic macrophages in HFD/obese mice. Diabetes. 2015;64(4):1120–1130.
- Matsumoto T, Takahashi N, Kojima T, et al. Soluble Siglec-9 suppresses arthritis in a collagen-induced arthritis mouse model and inhibits M1 activation of RAW264.7 macrophages. Arthritis Res Ther. 2016;18(1):133.
- Varchetta S, Mele D, Lombardi A, et al. Lack of Siglec-7 expression identifies a dysfunctional natural killer cell subset associated with liver inflammation and fibrosis in chronic HCV infection. Gut. 2016;65(12):1998–2006.
- Varchetta S, Lusso P, Hudspeth K, et al. Sialic acid-binding Ig-like lectin-7 interacts with HIV-1 gp120 and facilitates infection of CD4pos T cells and macrophages. Retrovirology. 2013;10:154.
- Kumagai E, Mano Y, Yoshio S, et al. Serum YKL-40 as a marker of liver fibrosis in patients with non-alcoholic fatty liver disease. Sci Rep. 2016;6:35282.
- European Association for the Study of the Liver; European Association for the Study of Diabetes; European Association for the Study of Obesity. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J Hepatol. 2016;64(6):1388–1402.
- Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2018;67(1):328–357.
- Watanabe S, Hashimoto E, Ikejima K, et al.; Japan Society of Hepatology. Evidence-based clinical practice guidelines for nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. J Gastroenterol. 2015;50(4):364–377.
- Ratziu V, Sanyal A, Harrison SA, et al. Cenicriviroc treatment for adults with nonalcoholic steatohepatitis and fibrosis: final analysis of the Phase 2b CENTAUR study. Hepatology. 2020;72(3):892–905.