
Abstract
Macrophage activation syndrome (MAS) is a severe, potentially life-threatening complication of systemic juvenile idiopathic arthritis (s-JIA). An immunological feature is the excessive activation and proliferation of T lymphocytes and macrophages. Massive hypercytokinemia is strongly associated with its pathogenesis, particularly the overproduction of interleukin (IL)-1, IL-6 and IL-18; interferon (IFN)-γ; and tumor necrosis factor (TNF)-α. Furthermore, heterozygous mutations in causative genes for primary hemophagocytic lymphohistiocytosis and in vivo exposure to highly elevated levels of IL-6 and IL-18 might induce natural killer cell dysfunction and decrease their numbers, respectively. A proper diagnosis is important to begin appropriate therapeutic interventions and change an unfavorable prognosis. The 2016 ACR/EULAR classification criteria for MAS have a high diagnostic performance; however, the diagnostic sensitivity for onset is relatively low. Therefore, careful monitoring of laboratory values during the course of MAS is necessary to diagnose it early in s-JIA. Further studies on the diagnosis and monitoring of disease activity using serum cytokine profile and a targeted cytokine strategy are required.
1. Introduction
Macrophage activation syndrome (MAS) is a severe, potentially life-threatening complication of rheumatic diseases, which is classified as a secondary form of hemophagocytic lymphohistiocytosis (HLH). MAS most often occurs in patients with systemic juvenile idiopathic arthritis (s-JIA) [Citation1]. s-JIA is characterized by arthritis and other systemic features such as spiking fever, salmon-colored skin rash, hepatosplenomegaly, generalized lymphadenopathy, and serositis [Citation2]. MAS is complicated in 7–14% of children with s-JIA [Citation3,Citation4]. Furthermore, subclinical or occult MAS may occur in another 30–40% of patients. MAS [Citation5], which develops at the onset of s-JIA in >20% of patients, may occur spontaneously or may be triggered by an infection or change in medication [Citation6–9].
MAS is characterized clinically by a non-remitting high fever, hepatosplenomegaly, cytopenia in all three blood cell lines, hepatic dysfunction, coagulopathy, central nervous system dysfunction, hyperferritinemia, and natural killer (NK) cell dysfunction [Citation1]. A pathognomonic feature is the presence of hemophagocytic macrophages found in the bone marrow [Citation1].
2. Pathogenesis of MAS
An immunological feature of MAS is the excessive activation and proliferation of T lymphocytes and macrophages. Massive hypercytokinemia is strongly associated with its pathogenesis. In particular, the overproduction of interleukin (IL)-1, IL-6, IL-12 and IL-18; interferon (IFN)-γ; and tumor necrosis factor (TNF)-α is closely associated with the pathogenesis of MAS complicating s-JIA () [Citation10]. Inadequate production of IL-10, a regulatory cytokine that counter-regulates IFN-γ, also might be related to the development of MAS in s-JIA [Citation11,Citation12].
Figure 1. MAS pathophysiology. The immunological feature of macropahge activation syndrome (MAS) is the excessive activation and proliferation of T lymphocytes and macrophages. Overproduction of interleukin (IL)-6 and IL-18, interferon (IFN)-γ, and tumor necrosis factor (TNF)-α, namely, cytokine storm, is closely associated with pathogenesis. In contrast, inadequate production of IL-10, a regulatory cytokine to counter-regulate IFN-γ, might be related to the development of MAS. Heterozygous mutations in causative genes for primary hemophagocytic lymphohistiocytosis related to granule-mediated cytolytic pathway and in vivo exposure highly elevated IL-6 and IL-18 levels and induced natural killer (NK) cell dysfunction and decreased the number of cells.
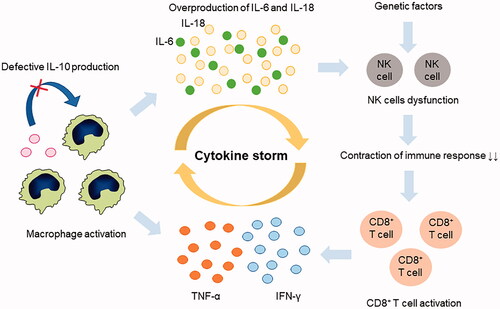
Furthermore, decreased NK-cell function, which is a characteristic feature of primary HLH (pHLH), has been observed in s-JIA and MAS [Citation13–17]. Reduced NK cytotoxic activity is associated with heterozygous mutations in causative genes for pHLH in some patients [Citation13,Citation18]. Moreover, in vivo exposure to highly elevated IL-6 and IL-18 might induce NK-cell dysfunction and their decrease, respectively [Citation19–23].
2.1. Genetic causes of impaired cytolytic function in s-JIA and MAS
pHLH is caused by mutations that affect the granule-mediated cytolytic pathway, including the PRF1, UNC13D, STX11, and STXBP2 genes in cytotoxic T lymphocytes (CTLs) and NK cells [Citation24–30]. The reduced cytolytic function of these cells leads to a persistent ongoing immune response and overproduction of inflammatory cytokines [Citation25,Citation26]. CTLs and NK cells perform their cytotoxic functions by releasing toxic granules containing perforin and granzymes. Once these cells are activated, cytotoxic granules are released into the immunological synapse between the antigen-presenting cells and the CTL and NK cells. Perforin forms a pore in the antigen-presenting cell, allowing granzyme B to enter the cytoplasm and trigger apoptosis.
Four steps are required for the release of cytotoxic granules: (1) sorting these proteins to the correct vesicle, (2) polarizing the vesicle to the immune synapse, (3) docking the vesicle with the plasma membrane, and (4) priming and fusing the vesicle () [Citation31]. Munc 13-4 encoded by the UNC13D gene is necessary for priming [Citation27]. Syntaxin 11, which is encoded by the STX11 gene, and syntaxin-binding protein 2, which is encoded by the STXBP2 gene, are essential for fusion [Citation29,Citation30].
Figure 2. Granule-mediated cytolytic pathway. Primary hemophagocytic lymphohistiocytosis is caused by mutations, including the PRF1, UNC13D, STX11, and STXBP2 genes, which affect the granule-mediated cytolytic pathway in cytotoxic T lymphocytes and (NK) cells. Four steps are required for the release of cytotoxic granules: (1) sorting these proteins to the correct vesicle, (2) polarizing the vesicle to the immune synapse, (3) docking the vesicle with the plasma membrane, and (4) priming and fusing the vesicle. Munc 13-4 encoded by the UNC13D gene is necessary for priming. Syntaxin 11 encoded by the STX11 gene and syntaxin-binding protein 2 encoded by the STXBP2 gene is essential for fusion of the vesicle.
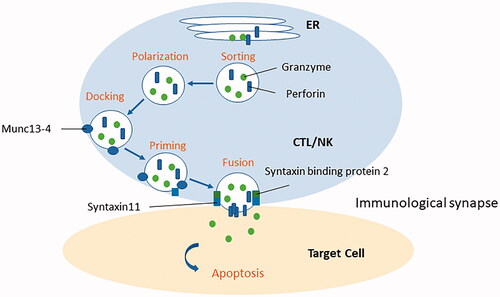
pHLH is classified into five subtypes. pHLH1-linked locus on chromosome 9q21.3 was reported in 1999 in four consanguineous families in Pakistan [Citation32]; however, to date, no causative genes have been identified. pHLH2 is caused by the mutations of the PRF1 gene-encoding perforin protein [Citation28]. pHLH3-5 is caused by mutations of the UNC13D, STX11, and STXBP2 genes [Citation27,Citation29,Citation30].
Mutations of the RAB27A gene-encoding Rab27a protein, a MUNC13-4 effector molecule, have been linked to the development of Griscelli syndrome type 2 [Citation33]. Mutations of the Lyst gene-encoding LYST protein, which is essential for lysosomal trafficking and protein sorting, have been identified to cause Chediak–Higashi syndrome [Citation34]. Mutations of the AP3B1 gene-encoding AP3βchain protein, which is essential for the trafficking from Golgi to granules, can cause Hermasly–Pudlak 2 syndrome [Citation35]. These three disorders can be complicated by pHLH with partial albinism.
Previous studies investigating mutations in the pHLH genes, including PRF1, UNC13D, STX11, STXBP2, and RAB27A, in patients with s-JIA have revealed that more than one-third of patients had monoallelic mutations [Citation13]. Furthermore, the frequency of rare protein-altering variants in UNC13D, STXBP2, and LYST were higher in patients with MAS (36%) as compared to those without (14%) [Citation18]. These results indicate that genetic mutations affecting granule-mediated cytolytic function might increase susceptibility to MAS in some patients with s-JIA, although further studies, including those with Japanese patients, are necessary to define the link between these mutations and MAS predisposition.
2.2. Acquired NK-cell dysfunction and IL-6 and IL-18 overproduction in s-JIA
Recent investigations have revealed that the innate proinflammatory cytokines IL-1, IL-6, and IL-18 play important roles in the pathogenesis of s-JIA [Citation10]. NK-cell development, number, and function are modulated by IL-6 and IL-18 [Citation19–23]. In an IL-6 transgenic mouse that developed MAS, elevated levels of IL-6 induced reductions in perforin and granzyme B expression in the NK cells without effects on degranulation that improved after treatment with IL-6 inhibition [Citation20]. Furthermore, similar changes have been observed in NK cells from patients with s-JIA [Citation20]. These findings indicate that IL-6 is involved in the amplification of inflammatory responses and suggest the role of chronic IL-6 overproduction in NK-cell dysfunction and MAS development.
The introduction of tocilizumab (TCZ), a humanized monoclonal antibody against the human IL-6 receptor, has brought about a paradigm shift in the treatment of s-JIA [Citation36,Citation37]. However, MAS still can develop in some patients with s-JIA during treatment with TCZ [Citation38–41]. This finding indicates that blocking IL-6 alone cannot completely prevent the onset of MAS.
We previously have reported that serum IL-18 levels were extremely elevated in patients with active s-JIA, and these further increased in patients with MAS [Citation42]. Increased serum IL-18 levels were specific to MAS with s-JIA because other forms of HLH are associated with lower serum IL-18 levels. Furthermore, we have identified two distinct groups of patients with s-JIA having specific clinical features on the basis of their IL-6 and IL-18 levels [Citation43]. Patients in the IL-6–dominant group (IL-18/IL-6 < 1000) appeared to have more severe joint disease, whereas those in the IL-18–dominant group (IL-18/IL-6 > 1000) were more likely to develop MAS [Citation44].
Moreover, recent studies have revealed a close association between IL-18 and the development of MAS [Citation45]. Canna et al. have identified a new monogenic autoinflammatory disease associated with gain-of-function mutations of the NLRC4 gene, an inflammasome sensor that causes a disease characterized by early-onset, recurrent MAS [Citation45]. Mutations of NLRC4 cause and trigger constitutive caspase-1 cleavage and increased production of IL-18 [Citation45]. The most characteristic immunological finding was extremely high IL-18 levels equal to those in patients with MAS and s-JIA, providing evidence that NLRC4 is particularly important in regulating IL-18 production and in further supporting the role of IL-18 as a predisposing factor for MAS. These findings indicate that IL-18 is another mediator in s-JIA and that its overproduction may be closely related to the development of MAS.
IL-18 is an interferon-γ-inducing factor mainly produced by activated macrophages that enhances the proliferation and activity of T cells and NK cells. IL-18 is the most effective at regulating NK-cell activity. Previous studies have reported that NK-cell dysfunction and a decreased number of cells were found in s-JIA [Citation16,Citation21–23]. A previous report has shown that exposure to high concentrations of IL-18 can induce NK-cell death [Citation21–23]. Furthermore, de Jager et al. have reported that impaired NK-cell function in s-JIA involves a defect in the IL-18 receptor β phosphorylation [Citation22]. These findings indicate that IL-18 exposure may decrease the number of NK cells and also impair their functions.
We previously have reported the case of an infant born to a mother with adult-onset Still’s disease (AOSD), presenting with transient impairment of NK-cell function associated with increased serum IL-18 levels, transmitting from the mother to the infant [Citation46]. Furthermore, infants born to mothers with AOSD can develop MAS and show extremely elevated levels of IL-18 [Citation47]. These findings indicate that the nonfunctional IL-18/NK-cell axis induced by high serum IL-18 levels may be closely associated with MAS development in s-JIA.
We have demonstrated that NK-cell activation by IL-18 is impaired in patients with active s-JIA [Citation23]. Furthermore, NK-cell activation is recovered by exogenous IL-18 stimulation in patients with s-JIA in whom serum IL-18 levels decreased after treatment began [Citation23]. Furthermore, NK-cell activation by IL-18 stimulation is negatively correlated with serum IL-18 levels [Citation23]. On the basis of these findings, exposure to high IL-18 levels may induce NK-cell exhaustion and secondary transient NK-cell dysfunction.
2.3. IFN-γ as the key effector cytokine in MAS
Elevated serum IFN-γ and IFN-γ-induced chemokine levels are significantly correlated with disease activity in pHLH, infection-associated secondary HLH, and MAS [Citation48–52]. Furthermore, the clinical symptoms of pHLH and MAS have been shown to be inhibited by anti-IFN-γ antibody treatments [Citation53,Citation54]. These findings indicate that IFN-γ plays a pivotal role in MAS as well as in HLH.
IL-10 is an immunosuppressive cytokine that counter-regulates IFN-γ. In patients with s-JIA, plasma IL-10 levels are lower as compared with proinflammatory mediators [Citation12]. Furthermore, B lymphocytes show decreased IL-10 production ex vivo and after in vitro stimulation [Citation12]. Furthermore, IL-10 polymorphisms are associated with decreased IL-10 activity [Citation11]. In a mouse model of MAS with repeated TLR9 stimulation, IL-10 receptor blocking increased disease severity [Citation55]. These findings indicate that defective IL-10 production and overproduction of IFN-γ contribute to the pathogenesis of MAS.
3. Clinical features of MAS
MAS is clinically characterized by sustained fever, hepatomegaly with liver dysfunction, splenomegaly, lymphadenopathy, and variable neurologic symptoms. Once MAS develops, fever patterns change from spiking to continuous, and the fever is usually high grade (>38.5 °C), prolonged, and unresponsive to anti-infection treatment. Hemorrhagic symptoms and respiratory distress are sometimes observed. The characteristic laboratory findings of MAS are leukocytopenia, thrombocytopenia, coagulation disorders including a decrease in plasma fibrinogen levels and increase in FDP and D-dimer levels, increased liver enzymes, elevated triglycerides, abnormally high ferritin levels, and NK-cell dysfunction. Hemophagocytosis found in bone marrow puncture is a hallmark of MAS, although hemophagocytes are not essential for the diagnosis of HLH/MAS and are found in patients with juvenile arthritis without overt MAS.
These abnormal findings are closely associated with the overproduction of inflammatory cytokines and can be explained by the known effects of key cytokines such as IFN-γ; TNF; and IL-1β, IL-6, and IL-18. shows the relationship between these and the characteristic clinical features of MAS.
Table 1. Relationship between cytokines and clinical manifestations of MAS.
Cytopenia results from both hemophagocytosis in the bone marrow and depression of hematopoiesis by IFN-γ, IL-1β, and TNF-α. Coagulopathy is associated with fibrin deficiency caused by liver dysfunction, and DIC develops as a result of IFN-γ and TNF overproduction. Liver dysfunction is associated with IFN-γ, which contributes to the development of cholestasis and causes liver damage. Hypertriglyceridemia is the consequence of the inhibition of lipoprotein lipase by TNF-α. Massive elevation of serum ferritin levels may be caused by IL-1β and TNF elevation and also reflects the activation of M2c-type macrophages. Elevation of urinary β2 microglobulin is induced by IFN-γ.
Monitoring the serum cytokine profile is useful for assessing the disease activity of s-JIA and MAS. However, it can be difficult to monitor cytokine levels in general hospitals. Thus, monitoring serum cytokine-inducible protein levels is useful to assess the disease activity of MAS ().
Table 2. Cytokine inducible proteins.
4. Diagnosis of MAS
4.1. Classification criteria
MAS is diagnosed using the 2016 ACR/EULAR classification criteria for MAS complicating s-JIA () [Citation56]. In this criteria, a febrile patient with known or suspected s-JIA is classified as having MAS if the patient has ferritin > 684 ng/L and at least two of the following laboratory abnormalities: platelets ≤ 181 × 109/mL, AST > 48 U/L, triglycerides > 1.76 mmol/L (156 mg/dL), and fibrinogen ≤ 3.6 g/L (360 mg/mL).
Table 3. 2016 ACR/EULAR classification criteria for MAS complicating s-JIA.
We previously have validated these classification criteria in Japanese children with MAS [Citation57]. These criteria had a high diagnostic performance; however, the diagnostic sensitivity for MAS onset was relatively low. Therefore, careful monitoring of laboratory values during the course of MAS, including platelet counts, plasma fibrinogen, FDP and FDP D-dimer levels, serum AST, LDH, and triglycerides, is necessary for the early diagnosis of MAS in s-JIA.
4.2. Differentiation from pHLH
It is important to differentiate MAS from pHLH, because treatment strategies and outcomes are significantly different. The MAS/HLH (MH) score uses the following items to make the differential diagnosis: age at onset, neutrophil count, fibrinogen, platelet count, and hemoglobin () [Citation58]. According to statistical weight, each item is assigned a score. The cut-off value of the MH score best in discriminating between MAS and pHLH is 60 (score range, 0–123 points), with a sensitivity of 91% and a specificity of 93% [Citation58].
Table 4. The MAS/HLH score.
4.3. Treatment with biologics
The clinical use of IL-1- and IL-6-blocking biologic agents have striking and long-lasting effects on s-JIA, even in patients receiving other therapies [Citation36,Citation37,Citation59–61]. However, despite the efficacy of these biologics, patients with s-JIA are still at risk for developing MAS [Citation38–41]. The rate of MAS complications in patients being treated with canakinumab and TCZ were 2.8 and 1.8 per 100 patients, respectively, which is similar to the rates reported for patients not being treated with biologics [Citation40]. Furthermore, these biologics can modify the clinical and laboratory features of MAS associated with S-JIA [Citation40,Citation41]. MAS classification criteria were less likely to classify patients diagnosed with MAS while being treated with canakinumab (77.1%) and TCZ (54.3%) because of an absence of fever or insufficient ferritin elevation, compared with the patients not receiving these biologics [Citation40]. These findings indicate that using the 2016 ACR/EULAR criteria for these patients may not be appropriate. Care must be taken to not underdiagnose MAS using these criteria.
4.4. Biomarkers for diagnosis
A proper diagnosis of MAS is important for beginning an appropriate therapeutic intervention and for changing an unfavorable prognosis. However, it often is difficult to distinguish MAS from s-JIA flares, especially in its early stages. Therefore, identifying a promising indicator of disease activity as well as a useful marker to predict the transition to MAS from acute-phase s-JIA is desired.
IL-18 levels in the serum are significantly increased in active s-JIA [Citation42,Citation62,Citation63]. Therefore, IL-18 increasingly has been recognized as a biomarker for s-JIA diagnosis and its disease activity [Citation63]. The levels of IL-18 increase more in patients with MAS [Citation42,Citation62,Citation63]. We have found that patients with s-JIA who have extremely high serum IL-18 levels (>47,750 pg/mL) were more likely to develop MAS [Citation44]. However, no significant differences existed in serum IL-18 levels in s-JIA patients with MAS whether measured before or during MAS. These findings indicate that IL-18 is causatively involved in the development of MAS, and serum IL-18 levels are useful for predicting MAS in s-JIA.
IL-18 is a well-known IFN-γ-inducing cytokine. Recently, high levels of free IL-18 (i.e., IL-18 not bound to IL-18-binding protein) have been found to indicate the risk for developing MAS [Citation62]. Increased free IL-18 and induction of IFN-γ might be closely related to the development of MAS in s-JIA. IFN-γ plays a central role in the pathogenesis of HLH and MAS. Serum levels of IFN-γ and IFN-γ-induced chemokines are markedly elevated in pHLH, in infection-related secondary HLH, and in MAS [Citation48–52]. These levels are significantly correlated with disease activity [Citation51,Citation52]. TNF-α is another key cytokine in the pathogenesis of HLH and MAS [Citation10]. Serum levels of sTNFR-II and sTNFR-II/I ratio reflecting TNF-α production are significantly elevated in patients with MAS [Citation64]. These levels also are significantly correlated with disease activity.
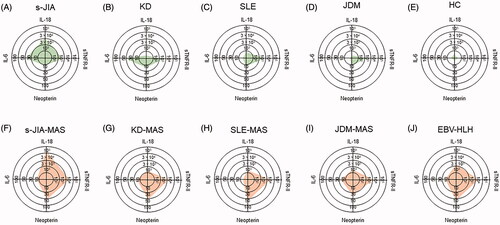
Serum cytokine levels with a radar chart in patients with MAS in different background rheumatic diseases and Epstein–Barr virus-associated HLH are shown in . The pattern of the cytokine profile is characteristic in each background. The massive increase of serum IL-18 levels is characteristic of s-JIA. Furthermore, we have compared the accuracy of serum biomarkers, including IFN-γ-related molecules and TNF-α-related molecules, for the diagnosis of MAS [Citation65]. Receiver operating characteristic (ROC) curve analysis has revealed that serum neopterin levels had the highest area under the ROC curve value, and neopterin was the most accurate biomarker for the diagnosis of MAS [Citation65]. From these findings, a combination of serum IL-18 and neopterin levels might be useful to diagnose MAS and to differentiate MAS from s-JIA flares or from other secondary HLH.
Figure 3. (A–J) Cytokine profiles with radar charts in patients with MAS in different background rheumatic diseases and Epstein–Barr virus associated HLH. Representative profiles of serum cytokines including, neopterin, IL-6, IL-18, and sTNF-RII are shown for in patients with MAS in different background rheumatic diseases and Epstein–Barr virus associated HLH. s-JIA: systemic juvenile idiopathic arthritis, KD: Kawasaki disease, SLE: systemic lupus erythematosus, JDM: juvenile dermatomyositis, HC: healthy controls, MAS: macrophage activation syndrome, EBV-HLH: Epstein–Barr virus associated hemophagocytic lymphohistiocytosis.
5. Treatment of MAS
The intravenous infusion of a high-dose glucocorticoid is the basic treatment for MAS. Methylprednisolone pulse therapy (30 mg/kg/time, 3 days as one course, one to two courses) is widely used as a first-line treatment. The intravenous infusion of dexamethasone palmitate (2.5–5.0 mg, every 12 h; half a dose for infants) also is effective. In addition, cyclosporine to suppress T cell activation and restrain mitochondrial permeability conversion by TNF-α is used widely (continuous intravenous infusion, 1–1.5 mg/kg/day) [Citation66,Citation67]. However, patients treated with corticosteroids and cyclosporine are at high risk for developing posterior reversible encephalopathy syndrome [Citation68,Citation69]; therefore, tightly controlling patient blood pressure is essential. In addition, anticoagulant therapy is needed. Apheresis therapy, including plasma exchange and leukocytapheresis, has been performed in refractory cases to corticosteroids and cyclosporine [Citation70–72].
Recently, rapid and dramatic benefits have been seen with the use of emaparumab, an anti-IFNγ monoclonal antibody, in patients with pHLH and MAS [Citation53,Citation54]. High-dose anakinra, an IL-1 receptor antagonist, has also been reported to be effective [Citation73]. Additional therapeutic protocols are needed.
6. Conclusions
MAS is a fatal complication of s-JIA. Its proper and timely diagnosis is essential to initiate appropriate therapeutic interventions and to improve unfavorable outcomes. The 2016 ACR/EULAR classification criteria for MAS complications in s-JIA have a high diagnostic performance; however, their diagnostic sensitivity for MAS onset is relatively low. Therefore, careful monitoring of laboratory values during the course of MAS is necessary for early diagnosis. Remarkable progress in the use of biologics, particularly against IL-1 and IFN-γ, has been seen in treatment. Additional effective and safe therapeutic strategies for MAS are desired.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Ravelli A, Grom AA, Behrens EM, et al. Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: diagnosis, genetics, pathophysiology and treatment. Genes Immun. 2012;13(4):289–298.
- Petty RE, Southwood TR, Manners P, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. 2004;31(2):390–392.
- Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child. 2001;85(5):421–426.
- Moradinejad MH, Ziaee V. The incidence of macrophage activation syndrome in children with rheumatic disorders. Minerva Pediatrica. 2011;63:459–466.
- Behrens EM, Beukelman T, Paessler M, et al. Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J Rheumatol. 2007;34(5):1133–1138.
- Minoia F, Davi S, Horne A, et al. Clinical features, treatment, and outcome of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a multinational, multicenter study of 362 patients. Arthritis & Rhematology. 2014;66(11):3160–3169.
- Stephan JL, Kone-Paut I, Galambrun C, et al. Reactive haemophagocytic syndrome in children with inflammatory disorders. A retrospective study of 24 patients. Rheumatology. 2001;40(11):1285–1292.
- Ravelli A. Macrophage activation syndrome. Curr Opin Rheumatol. 2002;14(5):548–552.
- Ramanan AV, Schneider R. Macrophage activation syndrome following initiation of etanercept in a child with systemic onset juvenile rheumatoid arthritis. J Rheumatol. 2003;30:401–403.
- Mellins ED, Macaubas C, Grom AA. Pathogenesis of systemic juvenile idiopathic arthritis: some answers, more questions. Nat Rev Rheumatol. 2011;7(7):416–426.
- Hersh AO, Prahalad S. Genetics of juvenile idiopathic arthritis. Rheum Dis Clin North Am. 2017;43(3):435–448.
- Avau A, Vandenhaute J, Malengier-Devlies B, et al. Insufficient IL-10 production as a mechanism underlying the pathogenesis of systemic juvenile idiopathic arthritis. J Immunol. 2018;201(9):2654–2663.
- Bracaglia C, Sieni E, Da Ros M, et al. Mutations of familial hemophagocytic lymphohistiocytosis (FHL) related genes and abnormalities of cytotoxicity function tests in patients with macrophage activation syndrome (MAS) occurring in systemic juvenile idiopathic arthritis (sJIA). Pediatr Rheum. 2014;12:53.
- Wulffraat NM, Rijkers GT, Elst E, et al. Reduced perforin expression in systemic juvenile idiopathic arthritis is restored by autologous stem-cell transplantation. Rheumatology. 2003;42(2):375–379.
- Grom AA, Villanueva J, Lee S, et al. Natural killer cell dysfunction in patients with systemic-onset juvenile rheumatoid arthritis and macrophage activation syndrome. J Pediatr. 2003;142(3):292–296.
- Villanueva J, Lee S, Giannini EH, et al. Natural killer cell dysfunction is a distinguishing feature of systemic onset juvenile rheumatoid arthritis and macrophage activation syndrome. Arthritis Res Ther. 2005;7(1):R30–R37.
- Put K, Vandenhaute J, Avau A, et al. Inflammatory gene expression profile and defective Interferon-γ and granzyme K in natural killer cells from systemic juvenile idiopathic arthritis patients. Arthritis Rheumatol. 2017;69(1):213–224.
- Kaufman KM, Linghu B, Szustakowski JD, et al. Whole-exome sequencing reveals overlap between macrophage activation syndrome in systemic juvenile idiopathic arthritis and familial hemophagocytic lymphohistiocytosis. Arthritis Rheumatol. 2014;66(12):3486–3495.
- Avau A, Put K, Wouters CH, et al. Cytokine balance and cytokine-driven natural killer cell dysfunction in systemic juvenile idiopathic arthritis. Cytokine Growth Factor Rev. 2015;26(1):35–45.
- Cifaldi L, Prencipe G, Caiello I, et al. Inhibition of natural killer cell cytotoxicity by interleukin-6: implications for the pathogenesis of macrophage activation syndrome. Arthritis Rheumatol. 2015;67(11):3037–3046.
- Shibatomi K, Ida H, Yamasaki S, et al. A novel role for interleukin-18 in human natural killer cell death: high serum levels and low natural killer cell numbers in patients with systemic autoimmune diseases. Arthritis Rheum. 2001;44(4):884–892.
- de Jager W, Vastert SJ, Beekman JM, et al. Defective phosphorylation of interleukin-18 receptor beta causes impaired natural killer cell function in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2009;60(9):2782–2793.
- Takakura M, Shimizu M, Yakoyama T, et al. Transient natural killer cell dysfunction associated with interleukin-18 overproduction in systemic juvenile idiopathic arthritis. Pediatr Int. 2018;60(10):984–985.
- Henter JI, Horne A, Arico M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–131.
- Filipovich AH. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology Am Soc Hematol Educ Program. 2009;2009(1):127–131.
- Sullivan KE, Delaat CA, Douglas SD, et al. Defective natural killer cell function in patients with hemophagocytic lymphohistiocytosis and in first degree relatives. Pediatr Res. 1998;44(4):465–468.
- Feldmann J, Callebaut I, Raposo G, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell. 2003;115(4):461–473.
- Stepp SE, Dufourcq-Lagelouse R, Le Deist F, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286(5446):1957–1959.
- Zur Stadt U, Rohr J, Seifert W, et al. Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to syntaxin 11. Am J Hum Genet. 2009;85(4):482–492.
- Zur Stadt U, Schmidt S, Kasper B, et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet. 2005;14(6):827–834.
- Usmani GN, Woda BA, Newburger PE. Advances in understanding the pathogenesis of HLH. Br J Haematol. 2013;161(5):609–622.
- Ohadi M, Lalloz MR, Sham P, et al. Localization of a gene for familial hemophagocytic lymphohistiocytosis at chromosome 9q21.3-22 by homozygosity mapping. Am J Hum Genet. 1999;64(1):165–171.
- Ménasché G, Pastural E, Feldmann J, et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet. 2000;25(2):173–176.
- Barbosa MD, Nguyen QA, Tchernev VT, et al. Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature. 1996;382(6588):262–265.
- Dell’Angelica EC, Shotelersuk V, Aguilar RC, et al. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol Cell. 1999;3(1):11–21.
- Yokota S, Imagawa T, Mori M, et al. Efficacy and safety of tocilizumab in patients with systemic-onset juvenile idiopathic arthritis: a randomised, double-blind, placebo-controlled, withdrawal phase III trial. Lancet. 2008;371(9617):998–1006.
- De Benedetti F, Brunner HI, Ruperto N, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367(25):2385–2395.
- Shimizu M, Nakagishi Y, Kasai K, et al. Tocilizumab masks the clinical symptoms of systemic juvenile idiopathic arthritis-associated macrophage activation syndrome: the diagnostic significance of interleukin-18 and interleukin-6. Cytokine. 2012;58(2):287–294.
- Yokota S, Itoh Y, Morio T, et al. Macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis under treatment with tocilizumab. J Rheumatol. 2015;42(4):712–722.
- Schulert GS, Minoia F, Bohnsack J, et al. Effect of biologic therapy on clinical and laboratory features of macrophage activation syndrome associated with systemic juvenile idiopathic arthritis. Arthritis Care Res. 2018;70(3):409–419.
- Shimizu M, Mizuta M, Okamoto N, et al. Tocilizumab modifies clinical and laboratory features of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Pediatr Rheumatol. 2020;18(1):2.
- Shimizu M, Yokoyama T, Yamada K, et al. Distinct cytokine profiles of systemic-onset juvenile idiopathic arthritis-associated macrophage activation syndrome with particular emphasis on the role of interleukin-18 in its pathogenesis. Rheumatology. 2010;49(9):1645–1653.
- Shimizu M, Nakagishi Y, Yachie A. Distinct subsets of patients with systemic juvenile idiopathic arthritis based on their cytokine profiles. Cytokine. 2013;61(2):345–348.
- Shimizu M, Nakagishi Y, Inoue N, et al. Interleukin-18 for predicting the development of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Clin Immunol. 2015;160(2):277–281.
- Canna SW, de Jesus AA, Gouni S, et al. Goldbach-Mansky R. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet. 2014;46(10):1140–1146.
- Shimizu M, Sakakibara Y, Kawano M, et al. Transient impairment of NK cell function in an infant born to a mother with adult-onset Still’s disease: perinatal effect of maternal IL-18. Clin Immunol. 2012;143(3):273–274.
- Shimizu M, Kizawa T, Kato R, et al. Macrophage activation syndrome in neonates born to mothers with adult-onset Still’s disease: perinatal effect of maternal IL-18. Clin Immunol. 2019;207:36–39.
- Henter JI, Elinder G, Soder O, et al. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood. 1991;78(11):2918–2922.
- Osugi Y, Hara J, Tagawa S, et al. Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood. 1997;89(11):4100–4103.
- Xu XJ, Tang YM, Song H, et al. Diagnostic accuracy of a specific cytokine pattern in hemophagocytic lymphohistiocytosis in children. J Pediatr. 2012;160(6):984–990.
- Bracaglia C, de Graaf K, Pires Marafon D, et al. Elevated circulating levels of interferon-γ and interferon-γ-induced chemokines characterise patients with macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Ann Rheum Dis. 2017;76(1):166–172.
- Mizuta M, Shimizu M, Inoue N, et al. Clinical significance of serum CXCL9 levels as a biomarker for systemic juvenile idiopathic arthritis associated macrophage activation syndrome. Cytokine. 2019;119:182–187.
- Locatelli F, Jordan MB, Allen C, et al. Emapalumab in children with primary hemophagocytic lymphohistiocytosis. N Engl J Med. 2020;382(19):1811–1822.
- Gabr JB, Liu E, Mian S, et al. Successful treatment of secondary macrophage activation syndrome with emapalumab in a patient with newly diagnosed adult-onset Still’s disease: case report and review of the literature. Ann Transl Med. 2020;8(14):887.
- Canna SW, Wrobel J, Chu N, et al. Interferon-γ mediates anemia but is dispensable for fulminant toll-like receptor 9-induced macrophage activation syndrome and hemophagocytosis in mice. Arthritis Rheum. 2013;65(7):1764–1775.
- Ravelli A, Minoia F, Davì S, et al. 2016 Classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative initiative. Arthritis Rheumatol. 2016;68(3):566–576.
- Shimizu M, Mizuta M, Yasumi T, et al. Validation of classification criteria of macrophage activation syndrome in Japanese patients with systemic juvenile idiopathic arthritis. Arthritis Care Res. 2018;70(9):1412–1415.
- Minoia F, Bovis F, Davì S, et al. Development and initial validation of the macrophage activation syndrome/Primary Hemophagocytic Lymphohistiocytosis Score, a diagnostic tool that differentiates primary hemophagocytic lymphohistiocytosis from macrophage activation syndrome. J Pediatr. 2017;189:72–78.
- Nishimura K, Hara R, Umebayashi H, et al. Efficacy and safety of canakinumab in systemic juvenile idiopathic arthritis: 48-week results from an open-label phase III study in Japanese patients. Mod Rheumatol. 2021;31(1):226–234.
- Ruperto N, Brunner HI, Quartier P, et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367(25):2396–2406.
- Lequerré T, Quartier P, Rosellini D, et al. Interleukin-1 receptor antagonist (anakinra) treatment in patients with systemic-onset juvenile idiopathic arthritis or adult onset Still disease: preliminary experience in France. Ann Rheum Dis. 2008;67(3):302–308.
- Weiss ES, Girard-Guyonvarc’h C, Holzinger D, et al. Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome. Blood. 2018;131(13):1442–1455.
- Mizuta M, Shimizu M, Inoue N, et al. Clinical significance of interleukin-18 for the diagnosis and prediction of disease course in systemic juvenile idiopathic arthritis. Rheumatology. 2020. DOI:https://doi.org/10.1093/rheumatology/keaa634.
- Shimizu M, Inoue N, Mizuta M, et al. Characteristic elevation of soluble TNF receptor II:I ratio in macrophage activation syndrome with systemic juvenile idiopathic arthritis. Clin Exp Immunol. 2018;191(3):349–355.
- Takakura M, Shimizu M, Irabu H, et al. Comparison of serum biomarkers for the diagnosis of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Clin Immunol. 2019;208:108252.
- Soriano ME, Nicolosi L, Bernardi P. Desensitization of the permeability transition pore by cyclosporin a prevents activation of the mitochondrial apoptotic pathway and liver damage by tumor necrosis factor-alpha. J Biol Chem. 2004;279(35):36803–36808.
- Mouy R, Stephan JL, Pillet P, et al. Efficacy of cyclosporine A in the treatment of macrophage activation syndrome in juvenile arthritis: report of five cases. J Pediatr. 1996;129(5):750–754.
- Gijtenbeek JM, van den Bent MJ, Vecht CJ. Cyclosporine neurotoxicity: a review. J Neurol. 1999;246(5):339–346.
- Kumar S, Rajam L. Posterior reversible encephalopathy syndrome (PRES/RPLS) during pulse steroid therapy in macrophage activation syndrome. Indian J Pediatr. 2011;78(8):1002–1004.
- Demirkol D, Yildizdas D, Bayrakci B, et al. Hyperferritinemia in the critically ill child with secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction syndrome/macrophage activation syndrome: what is the treatment? Crit Care. 2012;16(2):R52.
- Miyazono A, Abe J, Ogura M, et al. Successful remission induced by plasma exchange combined with leukocytapheresis against refractory systemic juvenile idiopathic arthritis. Eur J Pediatr. 2014;173(12):1557–1560.
- Kinjo N, Hamada K, Hirayama C, et al. Role of plasma exchange, leukocytapheresis, and plasma diafiltration in management of refractory macrophage activation syndrome. J Clin Apher. 2018;33(1):117–120.
- Meneghel A, Martini G, Amigoni A, et al. Case Report: life-threatening macrophage activation syndrome with fulminant myocarditis successfully rescued by high dose intravenous anakinra. Front Pediatr. 2020;8:635080.