
Abstract
SARS-CoV-2 virus has become a global health problem that has caused millions of deaths worldwide. The infection can present with multiple clinical features ranging from asymptomatic or mildly symptomatic patients to patients with severe or critical illness that can even lead to death. Although the immune system plays an important role in pathogen control, SARS-CoV-2 can drive dysregulation of this response and trigger severe immunopathology. Exploring the mechanisms of the immune response involved in host defense against SARS-CoV-2 allows us to understand its immunopathogenesis and possibly detect features that can be used as potential therapies to eliminate the virus. The main objective of this review on SARS-CoV-2 is to highlight the interaction between the virus and the immune response. We explore the function and action of the immune system, the expression of molecules at the site of infection that cause hyperinflammation and hypercoagulation disorders, the factors leading to the development of pneumonia and subsequent severe acute respiratory distress syndrome which is the leading cause of death in patients with COVID-19.
Graphical abstract
1. Introduction
The coronavirus family are viruses that cause a wide variety of clinical manifestations commonly respiratory in a broad range of hosts including mammals and birds [Citation1,Citation2]. The wide diversity of coronaviruses is influenced by several genetic factors, which contribute to the emergence of new variants and species that can adapt to different hosts and alter their tissue tropism causing in some cases severe repercussions [Citation3].
In Wuhan, China in 2019, a new coronavirus is detected for the first time, which is called Severe Acute Respiratory Syndrome associated Coronavirus 2 (SARS-CoV-2) in patients who presented with severe pneumonia [Citation4,Citation5]. Its origin could be in bats as analyses indicated that it shared ∼96.2% identity with the genome of RaTG13 a bat coronavirus [Citation6,Citation7]. Although it also shows to have a close relationship with CoV isolated from pangolin as it shares ∼91.02% similarity and despite the overall genome percentage being lower than RaTG13 it was detected to share ∼97.4% similarity which is the highest with respect to the receptor binding domain (RBD) sequence, this implies that it may be a good candidate of the origin of SARS-CoV-2 [Citation8,Citation9].
Coronaviruses are a continuing threat, to date seven species are known to have the ability to infect humans, HCoV-229E, HCoV-NL63, HCoV-OC43 and HCoV-HKU1 have mild impact on infected patients, while SARS-CoV, MERS-CoV and SARS-CoV-2 can cause severe disease [Citation10,Citation11]. All three coronaviruses are highly pathogenic but SARS-CoV-2 is differentiated by its high transmission rate which has resulted in a high number of cases worldwide [Citation12]. Symptoms are variable between patients can range from asymptomatic patients to patients with mild to severe symptoms, the most common symptoms detected in patients who presented with coronavirus disease 2019 (COVID-19) are: fever, dry cough, breathing difficulties, muscle pain, altered taste, loss of smell and headache [Citation13,Citation14].
The main route of transmission is person-to-person via respiratory droplets resulting from talking, coughing, sneezing or spitting [Citation15,Citation16]. Possible airborne transmission is still debated. The people could become infected by inhaling aerosols from infected patients that could be suspended in the air for a prolonged time due to their size and favorable environmental conditions, the aerosols could also spread over a longer distance due to airflow [Citation17,Citation18]. The likelihood of transmission in open air is very low compared to enclosed spaces where there is inadequate ventilation as respiratory aerosol accumulation may occur [Citation17,Citation19]. According to Wang et al. individuals with high viral load could shed up to 1.23 × 105 copies of viral RNA in a single cough, individuals with moderate viral load can generate up to 386 copies of viral RNA [Citation20].
Another mode of transmission may be by the oral-fecal route as the virus can be detected in the digestive system [Citation21]. Evidence of infection by contact with contaminated surfaces is scarce, however there is evidence of contamination in objects found in places close to infected people and in hospital environments that could last for a considerable time [Citation22–24]. In spite of this, several studies mention that the risk of this type of transmission is unlikely, although it is not ruled out that isolated cases could occur; this depends largely on hand washing and surface disinfection, since this reduces the probability of infection [Citation23,Citation25,Citation26]. It is worth noting that some studies where SARS-CoV-2 positive surface samples were obtained were unable to perform In vitro cell culture, suggesting that the surfaces could be contaminated with non-viable virus [Citation27–29].
It is essential to clarify the importance of hand washing since a recent study shows that SARS-CoV-2 averages a survival of 9 h on the skin, which would increase the risk of transmission by direct contact through the skin, the form to reduce the risk is by washing hands [Citation30].
The immune response is the body's reaction to defend itself against foreign biological agents, SARS-CoV-2 faces different physical and chemical barriers that try to limit its spread [Citation31]. The virus enters through the upper respiratory tract where the cells express cofactors and the virus entry receptor facilitating the infection of the cells, then it goes to the lungs and depending on the severity it can spread to other organs [Citation32,Citation33]. The immune response is made up of innate immunity and adaptive immunity, in SARS-CoV-2 infection the immune defense starts in the upper respiratory tract where there are immune barriers such as mucosal secretion that is made up of different antimicrobial proteins and antibodies that interfere with the primary infection [Citation34].
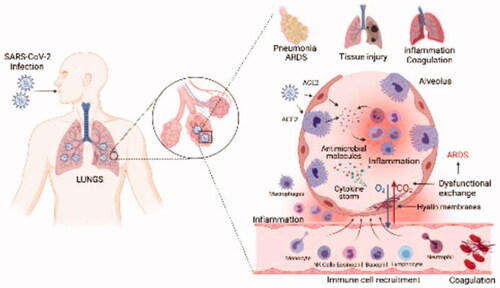
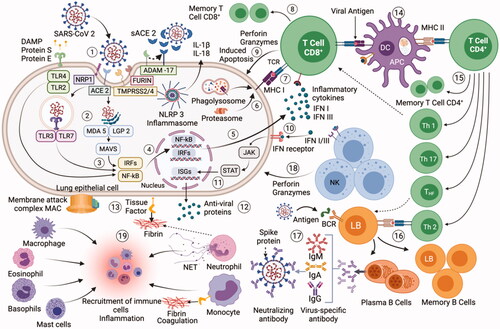
When SARS-CoV-2 infects host cells, a complex mechanism is activated that encompasses all components of the innate and adaptive immune system (). SARS-CoV-2 is recognized by immune receptors at different sites in the cell that activate signaling cascades for the production of pro-inflammatory cytokines and chemokines [Citation35,Citation36]. Several of these proteins send signals so that different immune cells such as: neutrophils, monocytes, eosinophils, NK cells, mast cells, basophils and lymphocytes are recruited to the site of infection and contribute to the elimination of the virus [Citation37,Citation38]. A determining response in the course of infection is the production of interferon, mainly Type I, if this response is early and robust it can correctly limit the SARS-CoV-2 replication [Citation36,Citation39]. The transition from innate to adaptive immunity is mediated by the presentation of antigens to T lymphocytes by specialized cells such as dendritic cells [Citation40]. SARS-CoV-2 activates the cellular and humoral adaptive immunity, where CD8 + T cells, CD4 + T cells and B cells act, B cells are responsible for the production of antibodies to neutralize the entry of SARS-CoV-2 into cells, the duration of immunological memory in recovered COVID-19 patients is not yet fully defined [Citation41,Citation42]. This network of immune interactions when they function correctly in patients with COVID-19 limits viral replication and the disease does not progress to its most severe and critical forms [Citation43].
Figure 1. Mechanism of SARS-CoV-2 interaction with the immune system: (1) infection and entry of SARS-CoV-2 into lung epithelial cells; (2) recognition of PAMP and DAMP by cell PRRs, activation of the inflammasome; (3) activation of signal translation pathways; (4) transcription of specific genes in the cell nucleus; (5) generation of proinflammatory cytokines and IFN; (6) antigen processing through the proteasome or by the phagolysosome; (7) antigen presentation by MHC I to CD8+ T cells; (8) clonal expansion, CD8+ T cells can differentiate into memory cells; (9) Active CD8+ T cells express antimicrobial molecules to eliminate the infected cell; (10) IFN-I and IFN-III can be recognized by IFN receptors on the cell surface; (11) signaling pathway is activated to promote ISG expression; (12) expression of more antiviral proteins is generated; (13) in the infected cell, complement system activation develops and TF expression is increased promoting fibrin-based coagulation; (14) dendritic cells present antigens through MHC-I and II to activate CD8+ and CD4+ T cells; (15) CD4+ T cells differentiate into different groups of helper and memory T cells; (16) B cells are activated through Th2 cells or by direct antigen recognition by a BCR receptor. B cells differentiate into plasma and memory cells; (17) Plasma cells produce antibodies specific for SARS-CoV-2; (18) NK cells can directly eliminate infected cells; (19) All immune cells can participate and be recruited to the site of lung infection and unleash inflammation and coagulation disorders leading to the more severe COVID-19 pathologies such as ARDS.
When the immune response is altered during SARS-CoV-2 infection, the outcomes can be harmful, several studies indicate that SARS-CoV-2 significantly dysregulates the mechanism of action of the immune system mainly in severe patients [Citation44–46]. First, SARS-CoV-2 can do the immune response deficient by suppressing the initial response mediated by type I and III interferon to facilitate viral shedding [Citation47]. In a second instance, the virus causes an uncontrolled immune response where there is the accumulation of cytotoxic molecules that instead of benefiting the elimination of the virus, contribute to the development of serious immunopathologies in patients such as loss of function and tissue damage [Citation48,Citation49]. The accumulation of cytotoxic molecules develops thanks to various actions of the immune response and the virus. SARS-CoV-2 could activate multiple immune receptors simultaneously () that work synergistically to express a high number of cytotoxic molecules [Citation50]. Another observed event is the abnormal recruitment of immune cells to the site of infection, the mechanism of action of these cells could be altered [Citation51]. The adaptive immune response is not adequate since lymphopenia develops and the production of antibodies is altered, in addition the regulatory T lymphocytes that maintain the homeostasis of the immune response decrease [Citation49,Citation52]. All these events contribute to an overexpression of cytotoxic molecules that trigger inflammation and coagulation problems in patients, leading the disease to its most serious disorders such as pneumonia and acute respiratory distress syndrome (ARDS) [Citation45]. In general, the immune system plays a key role in neutralizing the virus. There are different factors of both the virus and the immune response that influence the development of the disease as well as the patient's recovery [Citation53].
In this review we focus on the development of the immune response to SARS-CoV-2 infection from virus entry to the processes triggered by the organism to neutralize the infection, thus providing insights into each component of the immune system, immune cells involved in host defense and immunopathology in the development of COVID-19 disease.
2. Characteristics of the SARS-CoV-2 virus
SARS-CoV-2 belongs to the Coronaviridae family specifically to the Betacoronavirus (βCoV) genus [Citation54]. Its genome is composed of ∼30kb unsegmented positive sense single-stranded RNA, the viral genome encodes 16 non-structural proteins (nsp 1- 16), several accessory proteins (ORF3a, ORF 6, ORF 7a, ORF 7 b, ORF 8 and ORF 10) and 4 structural proteins known as: spike protein (S), membrane protein (M), nucleocapsid protein (N) and envelope protein (E) [Citation55,Citation56].
Protein S is of particular importance as it mediates the binding of the virus to the host cell and is also the main antigen that induces the production of neutralizing antibodies, which is ideal for the development of potential therapies against the virus [Citation57,Citation58]. S-glycoprotein is a protein with homo-trimeric structure belonging to the class I group of viral fusion proteins, each protomer is constituted by an S1 subunit possessing the receptor binding domain (RBD) and an S2 subunit possessing the membrane fusion machinery [Citation59,Citation60].
2.1. SARS-CoV-2 virus entry receptor: angiotensin-converting enzyme 2 (ACE2)
The first instance of viral infection progression is receptor recognition. The angiotensin-converting enzyme 2 (ACE2) is the main functional receptor recognized by SARS-CoV-2 virus to enter the host cell [Citation61–63]. ACE2 is a transmembrane enzyme that is part of the renin-angiotensin system (RAS) responsible for regulating the cardiovascular system, vasoconstriction, blood pressure, function of various organs, and body fluid homeostasis [Citation64,Citation65]. ACE2 has protective functions against diseases, it participates in the conversion of angiotensin I (Ang I) to angiotensin (1-9) (Ang-(1-9)) but its main function is to transform angiotensin II (Ang II) which is a potent vasoconstrictor into angiotensin (1-7) (Ang-(1-7)) a potent vasodilator, and functions as an antagonist of the negative effects of RAS by Ang II [Citation66,Citation67].
Ang-(1-7) exerts anti-inflammatory effects by binding to its receptor encoded by the Mas proto-oncogene (Mas receptor), negatively regulates the expression of proinflammatory cytokines such as: Interleukins (IL-6, IL-1β, IL-8), tumor necrosis factor alpha (TNFα) and monocyte chemoattractant protein 1 (MCP-1) [Citation68]. It also reduces the activation of signaling pathways: nuclear factor kappa B (NF-κB) and mitogen-activated kinases (MAPK) that are involved in the production of proinflammatory cytokines. In addition, Ang-(1-7) modulates extracellular signal-regulated kinase (ERK 1/2) that regulates the production of proinflammatory cytokines [Citation68,Citation69]. On the contrary, Ang II which is the main component of RAS, acts as a potent modulator of the immune system which enhances cytokine production by activating signal transduction pathways and induces the recruitment of proinflammatory cells [Citation68,Citation70]. Uncontrolled increase in their expression can lead to dysregulated inflammatory processes, alterations in blood pressure, cardiovascular dysfunction, systemic hypertension, pulmonary fibrosis and renal damage [Citation71].
Some studies show that neprilysin (NEP) in the RAS system has an important role in the pathogenesis of COVID-19 [Citation72,Citation73]. As known, NEP has important protective functions in pulmonary inflammation and fibrosis, its role in COVID-19 is not defined, it could contribute both to physiological processes and pathological conditions [Citation73].
NEP is expressed in the lungs and is another member of the RAS that can also induce Ang-(1-7) expression from Ang I or Ang-(1-9) so it could contribute with the conservation of Ang-(1-7) in COVID-19 [Citation73,Citation74]. It also has the ability to degrade bradykinin so it would help to decrease its inflammatory effects [Citation73,Citation75]. However, there are contradictory approaches that make known that NEP can further process Ang-(1-7) to smaller inactive peptides so the use of NEP inhibitors in combination with ACE2 stimulators would contribute to conserve Ang-(1-7) decreasing the negative effects of SARS-CoV-2 [Citation76]. Similarly, NEP can cleave peptides that are involved in the protection of the cardiovascular and pulmonary system, so the increase of its expression could contribute to the severity of patients with COVID-19 [Citation77]. Further studies are still needed to better understand the use of NEP as a therapeutic target against COVID-19.
2.2. Alteration of ACE2 receptor activity by the SARS-CoV-2 virus
In SARS-CoV-2 infection, there is a negative regulation of ACE2 activity that alters the balance of the RAS system and other substrates that are also processed by ACE2 such as apelin, dynorphin, neurotensin and bradykinin [Citation66,Citation78,Citation79]. RAS imbalance result in Ang II accumulation that could be involved in pathologies and injury to various organs in COVID-19 [Citation78,Citation80]. ACE2 is widely expressed in different organs and tissues including intestines, heart, kidneys, brain, testis and lungs specifically in type II alveolar epithelial cells, although it is also detected in type I cells [Citation81,Citation82]. Virus phagocytosed by macrophages and those that are free could target the different sites where ACE2 is expressed [Citation83]. This would explain how SARS-CoV-2 virus can cause multiple clinical manifestations such as respiratory, gastrointestinal, cardiovascular and renal problems observed in critically ill patients [Citation84,Citation85]. The difference of ACE2 expression levels in some organs is variable with respect to the vulnerability of SAR-CoV 2 infection. For example, the ileum is one of the organs where the highest ACE2 expression occurs but is not the most affected by the infection, in contrast there are organs that express to a lesser extent ACE2 and are affected by the virus [Citation82]. This demonstrates that there is a complex mechanism behind the infection and there may be other key components that participate and contribute to SARS-CoV-2 infection [Citation86].
2.3. Interaction of SARS-CoV-2 protein S with the ACE2 receptor
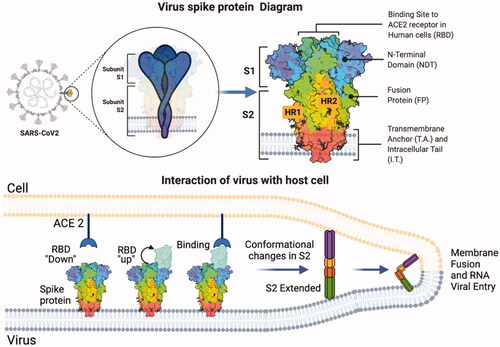
A key component in cellular receptor recognition is the RBD found on the SARS-CoV-2 S protein [Citation87,Citation88]. The RBD undergoes changes in its structural conformation, it can adopt a ‘down’ position which is a non-accessible state for immune evasion or an ‘up’ position, an active state in which binding determinants are exposed and interaction with the receptor develops [Citation59,Citation86,Citation89]. It binds specifically to the N-terminal peptidase domain of ACE2, once binding is accomplished additional factors such as proteases are required for viral and cell membrane fusion to occur [Citation86, Citation90]. The processing of protein S releases the fusion peptide, the S2 subunit undergoes conformational changes in its structure so that the fusion machinery facilitates viral and cell membrane binding giving way to the entry of its genetic material for replication within the cell [Citation91,Citation92] (). The viral RNA produced will be packaged by the formation of structural proteins and released from the cell by exocytosis, the viral progeny can continue to infect host cells [Citation93,Citation94].
Figure 2. Structural diagram of protein S and its interaction with ACE2 for membrane fusion. The SARS-CoV-2 spike protein has two subunits: S1 and S2. The RBD changes its structural conformation to an accessible position for the interaction with ACE2 to be established. Processing of protein S by cellular proteases releases the S2 subunit to change its structural conformation and facilitate viral and cell membrane fusion.
Cell surface heparan sulfate (HS) interacts with SARS-CoV-2 protein S through the RBD and is required for infection of cells through ACE2, HS acts as a co-receptor that enhances binding between protein S and ACE2 [Citation95,Citation96]. HS has multiple biological functions by binding to various proteins such as: cytokines, chemokines, growth factors and proteases, for example the interactions of HS with selectins and cytokines regulate the recruitment of immune cells to control inflammation, therefore HS is a target interesting therapeutic to modulate inflammation and reduce viral entry in COVID-19 [Citation95,Citation97].
Importantly, there is an RGD site in the RBD of the S protein of SARS-CoV-2 virus which is characterized by recognizing and binding to cellular integrins which could be a strategy to enhance virus entry or establish an alternate route of entry into cells [Citation98,Citation99]. One study observed that β1 integrins that are widely expressed in human lung cells are able to bind protein S and enhance virus entry [Citation100]. Integrins can regulate transforming growth factor beta (TGF-β) which is related to several clinical features of COVID-19, the use of integrin inhibitors could improve the status of patients and reduce SARS-CoV-2 entry into cells [Citation99,Citation101].
2.4. Alternate receptors that could be used by SARS-CoV-2 virus to enter cells
SARS-CoV-2 virus use alternate routes to infect host cells. The CD147 protein have the ability to interact with the S protein of SARS-CoV-2 [Citation102,Citation103]. T lymphocytes and other immune cells can be infected, but these types of cells express to a lesser extent ACE2 as opposed to CD147 which they express widely so the virus could use this protein as an alternate receptor to infect these types of cells [Citation103,Citation104]. CD147 is a transmembrane glycoprotein also known as Basigin, this protein has multiple functions in the regulation of the immune system and in the control of several pathologies such as: cancer, infectious diseases and inflammatory processes [Citation105,Citation106]. CD 209 (DC-SIGN) which is mainly expressed on dendritic cells and CD209L (L-SIGN) which is expressed on lung, liver and kidney cells, are receptors of the C-type lectin family that could also be able to recognize the SARS-CoV-2 virus and facilitate its entry into cells [Citation107,Citation108].
2.5. Factors with interaction between the host cell and the SARS-CoV-2 virus
2.5.1. Transmembrane serine protease 2 (TMPRSS2), cathepsin and furin
The entry of the virus into the cell requires the use of cellular proteases to activate the S protein. SARS-CoV-2 mainly uses the protease TMPRSS2 and Furin which would have an important role on the activation of the S protein [Citation62,Citation86]. Once the receptor is recognized, the S protein is cleaved at different positions, it possesses a furin cleavage site at the boundary between S1 and S2 (S1/S2) that pre-activates the S protein and a second proteolytic cleavage site at the S2 fusion peptide (S2') that is processed by cellular proteases as TMPRSS2 releasing the S2 subunit for membrane fusion to occur [Citation109,Citation110].
TMPRSS2 is a transmembrane protease that could be involved in different important physiological and pathological processes, however its main physiological role is still unknown [Citation111]. It is expressed in different sites such as: the respiratory tract, gastrointestinal tract, genitourinary system, ocular and olfactory system, there is evidence of high levels of protease expression in epithelial cells of the respiratory tract, lungs, trachea, bronchioles, nasal mucosa and pneumocytes of type II cells that are target cells for SARS-CoV-2 [Citation112]. On the surface of several types of cells, TMPRSS2 is co-expressed with ACE2 and Furin to act as the initial entry portal of the virus, it is important to note that ACE2 is not co-expressed with TMPRSS2 in all cells, so other proteases could participate in the virus entry [Citation62,Citation113]. The elevated expression of TMPRSS2 is related to the development and progression of several types of cancer, mainly prostate cancer, since the majority of individuals with cancer could be immunosuppressed and are more susceptible to contracting severe COVID-19 [Citation114].
Inhibition of TMPRSS2 and furin could be considered as a therapeutic approach against virus entry, however it should be kept in mind that furin is involved in the normal development of several processes in the organism [Citation115]. Furin is expressed in T lymphocytes and contributes to the processing of pathogen-fighting components [Citation116,Citation117]. It processes cytokines, chemokines and growth factors, in addition several infectious agents take advantage of its use to cleave fusion proteins and enter cells [Citation118]. Furin levels are often increased in certain conditions such as diabetes, obesity and aging, so it is speculated that it could be one of the reasons why they are a risk group for contracting COVID-19 [Citation119–121]. Importantly, the furin cleavage site could be one of the most important factors contributing with the high ability of SARS-CoV-2 virus to enter cells [Citation110]. It could also be one of the reasons explaining the efficient transmission of the virus since furin is detected in most organs and tissues of the human body and protein S could take advantage of its availability to pre-activate and establish early entry into respiratory cells [Citation110,Citation122].
SARS-CoV 2 prefers to activate its protein S through TMPRSS2 [Citation82]. However, when the TMPRSS2 expression is insufficient, the processing in S2' can also be carried out by the family of endosomal proteases cathepsins B and L [Citation62,Citation111]. But it should be noted that TMPRSS2 remains essential for virus entry into primary cells and viral spread unlike cathepsins which may be expendable [Citation62].
Cathepsin L has a more important role in the infection by SARS-CoV 2, in cells deficient in TMPRSS2 the virus is internalized in the cell by endocytosis, in the endosome the processing in S2' by cathepsin L is developed to produce membrane fusion [Citation111,Citation123]. Cathepsin L could improve the infection of SARS-CoV 2 since it increases its expression during chronic inflammatory states and in other diseases, the protease also contributes to degrade the extracellular matrix and increases inflammation [Citation124]. Cathepsin L and B are considered an important therapeutic target for the control of COVID-19., inhibiting them together with TMPRSS2 could be more beneficial to limit the infection [Citation125].
2.5.2. Transmembrane serine protease 4, 11 and 13 (TMPRSS4/11/13)
TMPRSS4 belongs to the same family as the serine protease TMPRSS2 and is involved in the cellular entry of several viruses such as influenza A virus and some coronaviruses [Citation126,Citation127]. Zang et al. evaluated SARS-CoV-2 infection in intestinal epithelial cells where they observed that TMPRSS4 promoted protein S cleavage and virus infectivity, they also demonstrated that a co-expression of TMPRSS4 and TMPRSS2 enhances SARS-CoV-2 entry in cell cultures [Citation128]. This protease is also expressed in the lungs and respiratory tract so the virus could use it to enhance its cellular infection [Citation129,Citation130]. Importantly, deregulation of TMPRSS4 can be correlated with cancer development [Citation131,Citation132]. In epithelial cells of lung adenocarcinomas, overexpression of TMPRSS4 occurs [Citation133], thus it could be implicated in the vulnerability and increased risk for severe COVID-19 events observed in lung cancer patients [Citation134–136]. Furthermore, TMPRSS4 may be related to several symptomatology observed in patients with COVID-19 as one study detected overexpression of the protease in the central nervous system and brain mainly in regions related to the sense of taste and smell [Citation137]. In general, there is evidence of the participation of TMPRSS4 in SARS-CoV-2 infection and its symptomatology. This protease has an important role in the entry of the virus so it could be considered as a therapeutic target for the control of COVID-19 however more studies are required [Citation133,Citation137].
Other transmembrane serine proteases such as: TMPRSS11D, TMPRSS11E, TMPRSS11F and TMPRSS13 can participate in the processing of the protein S of SARS-CoV 2, they are also expressed in cells and tissues that SARS-CoV 2 infects [Citation138–140]. TMPRSS13 is the protease that has a greater capacity to activate protein S although its efficiency is lower than TMPRSS2 and 4 [Citation113,Citation138]. Another study also detected that TMPRSS11A, which is a protease that is expressed in the epithelial cells of the respiratory tract, has the ability to process protein S [Citation141]. Kishimoto et al. Observed that the proteases TMPRRS11D and 13 have the greatest potential to improve the entry of the virus into cells [Citation140]. The different proteases that the virus can use to activate protein S can act together or instead of TMPRSS2, therefore, they contribute to improve the infectivity of SARS-CoV 2 in cells [Citation113]. These proteases are expressed in different cells and tissues where the virus can use them, this is one of the reasons that would help explain the high infectivity and viral pathogenesis of SARS-CoV 2 [Citation139].
2.5.3. Neuropilin-1 receptor
Neuropilin receptor-1 (NRP-1) acts as an entry factor of SARS-CoV-2 as it could enhance the interaction of the virus with ACE2 [Citation142,Citation143]. NRP-1 is a transmembrane glycoprotein that has affinity for substrates that are processed by furin such as protein S [Citation144]. NRP-1 can bind to different proteins, it is involved in the regulation of vacular permeability, cell proliferation, neuronal development, axon control and immune response, when it binds to the protein S of the virus in COVID-19 its signaling and functionality could be altered [Citation145].
Once protein S is processed at the furin cleavage site, a ‘C-end rule’ (CendR) site is formed that can interact with the b1 domain of NRP-1 to enhance SARS-CoV-2 infection [Citation146,Citation147]. NRP-1 is abundantly expressed on the surface of respiratory and olfactory epithelium, during infection the virus could enhance NRP-1 expression to favor its entry into cells [Citation142]. Due to its abundant expression in olfactory cells, NRP-1 could also be related to olfactory loss [Citation148]. It is also expressed in different immune cells and could influence altered inflammatory and coagulation activity in COVID-19 [Citation149,Citation150]. NRP-1 could be a potential therapeutic target to reduce SARS-CoV-2 entry. There are studies evaluating compounds with potential to interpose on NRP-1- protein S interaction [Citation151,Citation152].
2.5.4. Disintegrin and metalloprotease 17 (ADAM-17)
ADAM-17 is a key regulator of TNF-α, thanks to its proteolytic activity it processes multiple molecules such as: cytokines, receptors, growth factors, and cell adhesion molecules, so that its activity is important for various physiological processes [Citation153]. However, the alteration of its activity is related to multiple pathophysiological processes such as: inflammatory disorders, hypertension, kidney problems, lung problems, among others, therefore its inhibition is considered a therapeutic objective [Citation154,Citation155].
ADAM-17 can detach the ACE2 receptor from the cell surface and release it into the extracellular space [Citation67,Citation156]. TMPRSS2 also associates with ACE2 and could detach it as well as ADAM-17 leading to the development of competition between the two proteases for ACE2 processing [Citation157]. The mechanism by which ADAM-17 detaches ACE2 to the soluble space could presumably intercept virus entry and reduce its infectivity in cells [Citation158]. In fact, there are studies with soluble recombinant human ACE2 (rhsACE2) for the treatment of COVID-19, where it has the potential to limit the entry of the virus and protect from lung lesions in the early stages of infection [Citation159]. However, deletion of ACE2 from the cell surface could lead to abnormal processing of the systemic response which could lead to detrimental consequences in patients with COVID-19 [Citation160]. Furthermore SARS-CoV-2 infection could potentiate and upregulate ADAM-17 activity as it could influence virus entry and enhance its infectivity [Citation67,Citation156]. Accumulation of Ang II and activation of its Ang II type I receptor (AT1) can positively regulate ADAM-17 expression [Citation161,Citation162], as well as other factors that induce ADAM-17 activity such as: Toll-like receptors in response to pathogens [Citation162] and proinflammatory cytokines such as IL-1 β [Citation163].
2.6. Regulatory molecules of inflammation against the virus
2.6.1. Bradykinin
Bradykinin (BK) could be implicated in several of the pathologies observed in COVID-19. Several factors within the disease could contribute to an overexpression of bradykinins resulting in a ‘bradykinin storm’ [Citation164–167]. Key components of the Kinin-Kallikrein system (KKS) are expressed in alveolar lung cells targeted by SARS-CoV-2 and could therefore be affected during COVID-19 [Citation168]. KKS triggers the expression of BK and Lys-BK peptides which in turn by additional enzymes can be processed to generate desArg 9-BK (DABK) and Lys-desArg9-BK respectively [Citation169,Citation170]. The effects of the peptides are activated by recognizing and binding to two bradykinin receptors (RB1 and RB2) that are G protein-coupled, the ligand of RB1 is DABK and the ligand of RB2 is BK [Citation171,Citation172]. These peptides are related to the RAS system as ACE2 is responsible for inactivating DABK and ACE is responsible for inactivating BK [Citation169,Citation173]. KKS is involved in inflammatory processes, vasodilatation, hypotension, pain, vascular permeability, blood pressure, edema and the coagulation system [Citation174–176].
One of the pathologies that has discussed the involvement of bradykinins in COVID-19 is pulmonary edema or fluid accumulation [Citation165,Citation173]. Negative deregulation of ACE2 due to SARS-CoV-2 alters DABK degradation thereby inducing overexpression that increases vascular permeability and inflammation [Citation166,Citation169,Citation174]. BK also induces inflammation and edema so it could be involved in COVID-19 [Citation165,Citation166]. Inflammatory processes can stimulate RB1 and RB2 expression which increases DABK and BK levels triggering a further increase in inflammation [Citation166,Citation177,Citation178]. BK and DABK are also involved in cellular immune responses by stimulating the recruitment and activation of cells such as neutrophils and macrophages [Citation179,Citation180]. BK can even induce the production of proinflammatory cytokines as observed in colorectal cancer where it stimulates IL-6 expression [Citation181]. Additionally, it is important to mention that cough in COVID-19 could be induced by BK accumulation. Cough is generated when using ACE inhibitors or by direct inhalation of BK [Citation182,Citation183]. Karamyan suggests that apart from bradykinin there are other peptides that could have the same effects on the disorders observed in COVID-19 such as neurotensin and substance P, so it is likely that not only the bradykinin storm is involved in the pathology of SARS-CoV-2 but it is more likely that there is a ‘vasoactive peptide storm’ [Citation184].
On the other hand, it is important to note that there are studies that have shown that the normal expression of BK and its activity through its RB2 receptor can have protective effects, mainly cardiovascular [Citation185]. DABK and BK can be a potential therapeutic target for the control of COVID-19.
2.6.2. Apelin
Apelin (APLN) occurs in different isoforms that can be processed by ACE2 and have protective effects in multiple pathologies mainly at the cardiovascular level [Citation186,Citation187]. They function as a ligand of the apelin receptor (APJ) which is a G protein-coupled component [Citation188]. APLN regulates the RAS system as it can increase ACE2 expression and activity [Citation189,Citation190]. In addition, the APJ receptor can counteract the effects of Ang II by interacting with the AT1 receptor and blocking Ang II-AT1 binding [Citation189,Citation191]. The APLN-APJ system has an important role in the regulation of inflammatory processes. A decrease in APLN expression levels can stimulate the progression of inflammation and conversely an increase can protect tissues from inflammatory injury [Citation192,Citation193]. Notably, APLN has an inhibitory effect on the activation of the NF-κB signaling pathway [Citation194,Citation195]. In addition, apelin-13 may have an antagonistic role on NLRP3 inflammasome activation so it may have a protective role during COVID-19 [Citation195,Citation196]. The evidence that shows that APLN reduces inflammation and that counteracting the effects of ACE2 dysregulation observed in COVID-19, makes it a good option as a treatment in late stages of the disease, since at the beginning of the disease it could increase the entry of the virus by increasing the expression of ACE2 [Citation197].
2.6.3. Histamine
Histamine is a molecule that is synthesized and stored mainly in mast cells and basophils, it can be found in most tissues including the lungs [Citation198,Citation199]. It plays an important role in the development of inflammatory processes by stimulating or suppressing the production of proinflammatory cytokines and is a potent vasoactive molecule that increases vascular permeability [Citation200,Citation201]. Histamine can contribute both to inflammatory responses and to regulatory processes of the immune response thanks to its receptors, it also acts in physiological processes such as cell differentiation, proliferation and regeneration [Citation202].
Mast cells can be extensively activated in COVID-19 so they release large amounts of histamine which acts through their receptors found on the surface of different cells to stimulate the expression of proinflammatory cytokines and increase inflammation [Citation203]. In addition, histamine regulates activities of the immune response and is involved in the action of different cells such as T lymphocytes, B lymphocytes, mast cells and dendritic cells [Citation202,Citation204]. Elevated histamine expression could explain several clinical features observed in patients with COVID-19 [Citation205]. There are several studies that have evaluated the therapeutic potential of histamine receptor inhibitors for the treatment of COVID-19 where, demonstrating its therapeutic potential and therefore could be beneficial to use [Citation206,Citation207].
2.6.4. Endoplasmic reticulum aminopeptidases 1 and 2 (ERAP1 and ERAP2)
The role of ERAP1 and ERAP2 in the maintenance of COVID-19 could be important. These aminopeptidases participate in the regulation of the RAS system by transforming Ang II into Ang III and IV that together with their receptors produce anti-inflammatory effects [Citation208,Citation209]. The main feature of ERAP1 and ERAP2 focuses on an additional processing of the one carried out by the proteasome in the cytoplasm of the peptide structures of antigens [Citation210]. It aims to remove extensions of the N-terminal residue to optimize their size and prepare them in the assembly of their binding to the major histocompatibility complex class I (MHC-I) that will present the peptides to CD8+ T lymphocytes [Citation210,Citation211]. SARS-CoV-2 can stimulate the expression of ERAP1 and ERAP2 isoforms which could indicate their possible involvement in the processing of antigenic peptides of the virus [Citation212]. Even, Stamatakis et al. analyzed the processing of SARS-CoV-2 glycoprotein S1 by aminopeptidases resulting in smaller antigenic peptides that could be loaded and presented by MHC-I [Citation213]. ERAP1 expression regulates inflammatory processes and modulates innate and adaptive immune responses. Once any pathogen is detected it can stimulate excessive immune responses [Citation214].
2.7. Innate immune response to SARS-CoV-2 virus
The innate immune response is the first line of defense against a pathogen entering the body of an individual, this response could be involved in the development of the ‘cytokine storm’ or hyperinflammation syndrome that manifests in patients with severe COVID-19 [Citation215]. The innate immune response consists of physical and chemical barriers, in addition to the involvement of several immune cells that are primed to respond to various pathogens including SARS-CoV-2 virus [Citation216,Citation217] (). The presence of SARS-CoV-2 viral proteins and RNA acting as pathogen-associated molecular patterns (PAMPs) and substances generated in response to stress or cellular damage acting as damage-associated molecular patterns (DAMPs) are detected by pattern recognition receptors (PRRs) equipped on different host cell types that stimulate signaling cascades for the production of antiviral molecules [Citation215,Citation218].
Studies on activated immune receptors in COVID-19 are limited, receptor expression patterns might differ between mild and severe patients [Citation50]. SARS-CoV-2 can be detected by several PRRs () such as Toll-like receptors (TLRs) as there is evidence in patients with severe COVID-19 that it is positively regulated by TLR7 receptor expression that detects its viral RNA in the endosome [Citation50,Citation219], TLR4 receptor that is activated upon detection of the S protein [Citation50,Citation220], TLR 2 is activated upon recognition of the E protein of the virus [Citation221]. Both TLR2 and TLR4 can recognize viral glycoproteins and different cell damage molecules and are expressed on the cell surface [Citation220,Citation222]. The TLR 3 receptor that recognizes double-stranded RNA (dsRNA) in the endosome is activated in the presence of SARS-CoV 2 and data indicate that TLR8 is not activated or its levels decrease during COVID-19 [Citation50,Citation223]. Low TLR 3 expression and increased TLR 4 expression is associated with increased severity in COVID-19 patients [Citation223].
TLR4 could be one of the main receptors responsible for the recognition of SARS-CoV-2 PAMPs and one of those responsible for triggering the development of inflammatory processes [Citation224,Citation225]. TLR4 triggers the activation of signaling pathways that mediate the production of IL-6 and TNF-α which are two of the main cytokines detected in severe patient with COVID-19 [Citation220]. There is a strong interaction between SARS-CoV-2 protein S and TLR 4 [Citation220,Citation224]. Even, Shirato and Kizaki observed that a potent activation of NF-κB and MAPK signaling pathway occurs when TLR4 was stimulated with SARS-CoV-2 S1 protein in macrophages which triggers a robust inflammatory response [Citation225].
SARS-CoV-2 virus can also activate RIG-like receptors (RLRs). In lung epithelial cells with SARS-CoV-2 infection, melanoma differentiation-associated protein 5 (MDA5) is activated [Citation226,Citation227]. Yin and co-workers also observed that the receptor known as laboratory of genetics and physiology 2 (LGP2) belonging to RLRs acts in SARS-CoV-2 recognition, both MDA5 and LGP2 drive the production of type I interferon (IFN) and type III IFN in response to SARS-CoV-2 infection [Citation227]. MDA5 and LGP 2 are cytoplasmic receptors that can recognize dsRNA [Citation222,Citation228]. Additionally, the retinoic acid-inducible gene I (RIG-I) can restrict virus replication in lung cells mainly in the early phase of infection by recognizing virus-released positive-stranded RNA in the cytoplasm [Citation229]. Nucleotide-binding oligomerization domain-containing protein 1 (NOD1) belonging to the NOD-like receptor (NLR) family could similarly be activated in SARS-CoV-2 infection [Citation227].
The NOD-like inflammasome or NOD-like receptor containing pyrin domain 3 (NLRP3) can also be activated in SARS-CoV-2 infection [Citation50,Citation230]. SARS-CoV 2 N protein can induce NLRP3 activation and promote inflammatory responses [Citation231]. NLRP3 plays an important role in the pathogenesis of SARS-CoV-2 so blocking its activation could be a potential therapeutic target for treatment against COVID-19 [Citation230,Citation232]. NLRP3 contributes to inflammatory processes as it triggers the activation and release of IL-1β and IL-18 through the action of caspase-1 which processes proIL-1β and proIL-18 converting them to their active form [Citation233]. Another role of caspase-1 is that it can induce the development of pyroptosis through the processing of gasdermin D protein (GSDMD) which induces membrane pore formation [Citation233,Citation234]. A recent study detected that in embryonic-like stem cells and hematopoietic stem cells NLRP3 activation occurs after interaction between SARS-CoV-2 protein S and ACE2, elevated NLRP3 activation can cause tissue and organ damage by subjecting different cell types to their elimination by pyroptosis [Citation235].
2.7.1. Signaling cascades in SARS-CoV2 virus activation
Once SARS-CoV2 is recognized by PRRs a signaling cascade occurs that activates protein complexes that induce the expression of proinflammatory chemokines, interferons and cytokines as a defense mechanism to clear the pathogen from host cells [Citation218,Citation219]. PAMP-bound receptors interact with modulatory proteins such as myeloid differentiation primary response protein 88 (MyD88) that interacts with TLR4, TLR7 and TLR8, also the TIR domain-containing adaptor-inducible interferon-β (TRIF) that interacts with TLR3 and TLR4, mitochondrial antiviral signaling protein (MAVS) that interacts with MDA5 and RIG-I [Citation236]. These proteins trigger the activation of signal transcription factors such as NF-kβ and interferon regulatory transcription factors 3 and 7 (IRF3 and IRF7), the factors translocate to the cell nucleus to induce the expression of IFN and proinflammatory cytokines [Citation47,Citation236]. Type I and type III IFN molecules upon binding to their respective interferon receptors on lung epithelial cells which are: IFN-α/β receptor (IFNAR) and IFN-λ receptor (IFNLR) activate the janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway that is involved in the production of proinflammatory cytokines and activation of IFN-induced genes (ISGs) that further express antiviral proteins [Citation47,Citation227,Citation237] (). IFN- γ that is generally produced by T lymphocytes and NK cells, binds to IFN- γ receptor (IFNGR) on cells and activates JAK/STAT signaling pathway [Citation238]. Importantly, Ang II promotes its actions through activation of the JAK/STAT pathway contributing to the expression of proinflammatory cytokines, in addition the IL6/JAK/STAT3 signaling pathway further potentiates inflammatory processes in COVID-19 [Citation239,Citation240].
2.7.2. Cytokines and chemokines in COVID-19
Cytokines are characterized by being in a dysregulated state where there is an increase in their expression leading to a stage of hyperinflammation or also considered as ‘cytokine storm’ that can lead to ARDS in patient with COVID-19 [Citation51,Citation241]. Cytokine production increases as a function of severity in COVID-19 patients, elevated production of IL-6, IL-2, IL-7, IL-10, IL-15, IL-17, IL-18, IL-1α, IL-1β, granulocyte and macrophage stimulating factor (GM-CSF) and TNF-α was observed, furthermore, positively regulated genes of NF- κB pathway were detected in severe patients [Citation241–244]. describes some characteristics of the main antiviral proteins involved in severe COVID-19. Analyses in Bronchoalveolar Lavage Fluid (BALF) have also detected elevated expression of chemokines such as: CC chemokine ligands (CCL2 (MCP-1), CCL7 (MCP-3), CCL3 also known as macrophage inflammatory protein 1 alpha (MIP-1α) and CCL4 (MIP-1β)) and CXC chemokine ligands (CXCL1, CXCL2, CXCL8 (IL-8), CXCL17 and CXCL10 also known as interferon gamma-induced protein-10 (IP-10)) [Citation272,Citation273]. Similarly in blood samples from patients with COVID-19 elevated signatures of chemokines such as MCP-1, MCP-3, CCL8 (MCP-2), CXCL2, CXCL8, CXCL9, IP-10, CXCL16, MIP-1α and MIP-1β were obtained, several of these chemokines are chemoattractants of immune cells such as monocytes, neutrophils, T cells and NK cells [Citation46,Citation274,Citation275]. The cytokines and chemokines produced attract a greater number of effector cells which increases the development of inflammation causing damage in the lungs of patients with COVID-19 [Citation260].
Table 1. Influence of the main proinflammatory cytokines involved in the development of severe COVID-19.
The biological effects that cytokines and chemokines develop are regulated by cellular receptors that stimulate different signal translation pathways for the correct functioning of the immune response, due to the dysregulation in their expression during COVID-19, they contribute to the immunopathogenesis of the disease [Citation276]. In COVID-19, the increase in the expression levels of the different chemokine receptors (CCR and CXCR) such as: CCR1, CCR2, CCR3, CCR5, CCR6, CCR7, CCR10, CXCR1, CXCR2, CXCR3, CXCR4 and CXCR6, is associated with unfavorable prognoses in the majority of infected patients since chemokines attract a greater number of immune cells and stimulate the production of antimicrobial molecules [Citation38].
During infection, cytokines such as: IL-6, IL-2, TNF-α and IL-1 β interact with their receptors in a positive feedback loop that stimulates greater production of pro-inflammatory genes through the activation of different pathways of signaling such as NF-kβ and JAK/STAT which contributes to the severity of patients [Citation276–278]. Several studies have evaluated the possibility of using cytokine and chemokine antagonists as therapeutic strategies to treat the disease [Citation37,Citation277,Citation278].
2.7.3. Interferons
IFN type I, II and III are important in the innate immune response to limit the spread and replication of viral pathogens, in patients with COVID-19 there is a delayed response in IFN-I and IFN-III production which may be due to strategies of the virus to evade the innate immune response at the onset of infection [Citation227,Citation244,Citation263]. The levels of IFN-I represented by (IFN-α and IFN-β) and IFN-III represented by (IFN-λ1-4) in COVID-19 vary according to the time of infection and patient status, some studies have reported a reduced INF-I and INF-III response at the onset of SARS-CoV-2 infection that was related to patients who later presented severe disease pictures [Citation244,Citation274,Citation279]. In BALF from patients who developed severe COVID-19, IFN-I and III expression increases and could influence the immunopathology of infected patients [Citation266,Citation272]. In peripheral blood immune cells from patients with severe COVID-19, IFN-I expression is decreased compared to mild or moderate disease, IFN-β is not detected and there is altered IFN-α production which could be the result of cell migration to sites of infection such as the lung where they contribute to patient severity by increasing IFN-I expression [Citation279]. A recent study by Banerjee et al. demonstrated that the antiviral response to SARS-CoV-2 infection induces a predominant IFN-I response in in vitro analysis and in mild cases of COIVD-19 which may be sufficient to limit viral replication, whereas in severe patients this response is delayed, possibly harmful and could contribute to patient severity [Citation264].
There is conflicting information regarding IFN-λ which could be used as a potential therapy in early stages of infection as it has antiviral activity, acts on mucosa, generates less damaging proinflammatory responses than the other IFNs, its receptor is limitedly expressed, has action on immune cells and could restrict SARS-CoV-2 replication in bronchial epithelial cells [Citation267,Citation268,Citation280]. However, IFN-λ can reduce tissue repair and cell proliferation in the lung, being a factor that would be involved in compromising the epithelial barrier to secondary infections [Citation266,Citation281]. Secondary infections could be related to the severity of patients with COVID-19 as the SARS-CoV-2 alone could not generate excessive cytokine activation and one possibility is that patients triggering a severe disease reaction may be due to co-infection with other microorganisms such as bacteria and fungi that would contribute with the activation of multiple sets of immune receptors [Citation50].
Type II IFN consisting solely of INF-γ has shown a decreased expression pattern in patients with mild to moderate disease and an increase in patients with severe COVID-19 [Citation242,Citation244]. SARS-CoV-2 is generally sensitive to IFN activity but still there are high viral loads at symptom onset so the virus could has several strategies to suppress this immune barrier [Citation47,Citation282]. SARS-CoV-2 expresses several proteins that have the ability to inhibit IFN production such as ORF 6 protein which is one of the proteins that induces a major blockade of IFN expression, can inhibit STAT translocation to the cell nucleus which limits ISG expression [Citation283,Citation284]. ORF3a protein can inhibit stimulator of interferon genes (STING) which is important in innate immune response and IFN I production [Citation285]. ORF 9 b from SARS-CoV-2 similarly has the ability to inhibit type I and III IFN expression as it alters the action of several molecules involved in various signaling pathways for IFN production [Citation286].
2.8. Immune cells in response to SARS-CoV-2
2.8.1. Monocytes-macrophages
Macrophages play an important role in the body's defense against a pathogen by producing antimicrobial molecules, performing phagocytosis and acting as antigen presenting cells (APCs), alterations in their activity can lead to complications in various pathologies including COVID-19 [Citation287,Citation288]. In patients with severe COVID-19, the number of inflammatory monocyte-derived macrophages increases extensively in the lungs [Citation248,Citation289,Citation290], while the population of alveolar macrophages that produce anti-inflammatory responses decreases [Citation290]. When lung infection occurs or due to inflammatory conditions an increase in macrophage numbers is triggered by the recruitment of monocytes to the lung where they differentiate into macrophages [Citation288]. Both IL-6 and GM-CSF which are widely expressed in COVID-19 play an important role in the differentiation of monocytes to macrophages [Citation248]. In addition, the analyzes show overexpression of several chemokine-encoding genes in patient with COVID-19 capable of recruiting an increased number of inflammatory monocytes [Citation255,Citation291].
Uncontrolled macrophage activation and proliferation that generates elevated expression of proinflammatory cytokines is known as macrophage activation syndrome (MAS) [Citation254,Citation292]. Some studies have suggested that in patients with severe COVID-19, MAS may be a major contributing cause of exaggerated proinflammatory cytokine production and may be related to coagulation abnormalities [Citation288,Citation289]. The proinflammatory cytokines that are mainly produced in MAS are GM-CSF, TNF-α, IL-6 and IL-1β which are widely detected in COVID-19, furthermore other factors that could suggest the involvement of MAS in patients who are infected with SARS-CoV2 is the increased levels of ferritin, dimer D and C-reactive protein (CRP) [Citation291,Citation293,Citation294]. Another important aspect to discuss is the dysregulation of monocytes and macrophages in coagulation as various stimuli such as PAMP, DAMP and proinflammatory cytokines activate tissue factor (TF) expression in monocytes although it is also expressed in endothelial cells inducing an elevated fibrin-based coagulation state, SARS-CoV-2 could initiate an overproduction of TF elevating coagulation processes in patients [Citation289,Citation295].
Macrophages and monocytes could also be directly infected by SARS-CoV-2, analyses performed on alveoli and lung lymphoid tissue from patients killed by COVID-19 have detected the ACE2 receptor and viral particles in this cell type [Citation296]. Although it could also be due to the fact that infected epithelial cells can be taken up by phagocytosis of macrophages introducing viral particles into the cell, the expression of ACE2 on macrophages could be stimulated by inflammatory signals [Citation289,Citation297]. Recent studies observed that SARS-CoV-2 can infect monocyte-derived macrophages via ACE2 but with ineffective replication, despite this SARS-CoV-2 stimulated elevated cytokine and chemokine expression in these cells suggesting that although there is no productive infection, macrophages play an important role in the pathogenesis of COVID-19 [Citation256,Citation298]. SARS-CoV-2 can activate several PRRs in macrophages such as TLR7, TLR4 receptors, Fcγ receptors (FcγR) sensing anti-protein S IgG immune complexes and NLRP3 activation is also induced, all these factors contribute with elevated macrophage activity in the production of proinflammatory cytokines [Citation255,Citation289].
Monocytes and macrophages have a wide variety of functions, macrophages can be found in two phenotypes: M1 macrophages that trigger inflammatory responses and M2 macrophages that trigger anti-inflammatory responses and contribute to tissue repair, altering the balance between these two types can lead to serious complications [Citation299]. Patients with a high SARS-CoV 2 viral load have a predominant infiltration of M1 macrophages that favors the pathogenesis of the virus [Citation300] Monocytes are important for pathogen control and production of cytokines such as IL-6, SARS-CoV-2 deregulates the activity and quantity of blood monocytes to enhance its pathogenesis [Citation265,Citation288,Citation290]. Circulating monocytes in severe COVID-19 exhibited a mixed M1/M2 phenotype that could promote both inflammation and fibrosis with the aim of tissue repair although it could result in pulmonary complication in COVID19 [Citation301]. In general SARS-CoV-2 promotes monocyte trafficking to the lung and uncontrolled accumulation of macrophages which is associated with the severity level of infected patients [Citation302]. Macrophages could play an important role in the development of COVID-19-associated pneumonia and ARDS [Citation254,Citation293].
2.8.2. Neutrophils
Neutrophils are among the first cells to be recruited during an infection, they fulfill several important functions in the innate immune response such as phagocytosis, generation of reactive oxygen species (ROS), activation of other immune cells, release of antiviral proteins and the generation of neutrophil extracellular traps (NET) [Citation303,Citation304]. The number of circulating neutrophils in patients with COVID-19 increases (neutrophilia) and correlates with patient severity [Citation305–307]. Neutrophil recruitment to the site of infection in the lungs also increases [Citation308,Citation309]. Their accumulation favors the inflammatory state, lung damage and coagulation problems in the lungs of patients with COVID-19 [Citation308]. ACE2 controls neutrophil accumulation, deregulation of ACE2 in COVID-19 favors their infiltration at sites of infection and enhances neutrophilic [Citation310].
NETs are extracellular DNA networks formed of chromatin and antimicrobial proteins, neutrophils release them in a process known as NETosis with the aim of neutralizing pathogens and preventing their spread, but if dysregulation in their expression occurs it can cause detrimental effects [Citation303,Citation311]. In SARS-CoV-2 infection neutrophils in plasma, tracheal aspirate and lung increase NET production levels [Citation309,Citation312,Citation313]. Excessive production influences the severity of COVID-19 patients by contributing to inflammation, coagulation abnormalities and lung tissue damage [Citation260,Citation307]. When NET expression occurs, other molecules are released such as neutrophil elastase, histones, granular proteins, cathepsin G, among others that act as DAMPs and activate several PRRs inducing the production of proinflammatory cytokines leading to additional tissue damage [Citation310]. One of the most important by-products of neutrophils is neutrophil elastase. This molecule is a protease that could potentiate virus entry, because it could also process SARS-CoV-2 protein S [Citation307,Citation311]. Elastase imbalance is related to acute lung injury and increased inflammation because it can induce damage to the alveolar-capillary barrier [Citation308].
Veras et al. observed that NET production in COVID-19 can be directly activated by SARS-CoV-2 as it can infect neutrophils, NET can also develop by several factors including activation of PRRs such as TLR7 which is associated with NET formation and also several cytokines and chemokines produced upon infection [Citation309]. IL-1β which is widely expressed in COVID.19 is a key inducer in NET formation, macrophages produce IL-1β through NLRP3 inflammasome activity contributing with NET formation and likewise NET could drive macrophage activation and NLRP3 to increase IL-1β expression in a positive feedback loop that may eventually have detrimental effects in COVID-19 patients [Citation260,Citation261]. IL-17 which is expressed in COVID-19 can promote neutrophil recruitment to site of infection where they contribute with the inflammation [Citation314], IL-17 can also promote NET formation [Citation315]. CCL20 which is elevated in COVID-19 is a potent inducer of NET [Citation316]. NET can drive thrombus formation in blood vessels as it can form aggregates, stimulate platelet accumulation and activate the complement system [Citation310,Citation317].
2.8.3. Eosinophils
Eosinophils are potent proinflammatory cells that in their granules contain cytotoxic molecules such as: eosinophil peroxidase, major basic protein-1, eosinophil cationic protein and eosinophil-derived neurotoxin [Citation318,Citation319]. These molecules could neutralize SARS-CoV-2 so the virus could alters eosinophil response and levels to maintain its spread [Citation320]. Most patients with COVID-19 have a decreased eosinophil count (eosinopenia) which is related to patient severity [Citation321–323]. Several studies have included eosinophil count in algorithms to predict severity in patients with COVID-19 [Citation324], although other factors may influence it so it is not definitive in determining the presence and pathology of SARS-CoV-2.
The mechanism by which SARS-CoV-2 induces eosinopenia is unclear, but it could be due to several factors such as a hyperinflammatory state that could modulate the eosinophil response, a blockade of eosinophil production and release from the bone marrow, and could also be due to decreased survival of eosinophils in the peripheral circulation [Citation318,Citation324]. Although some studies has also suggested that it could be due to functional depletion by eosinophils acting to clear SARS-CoV-2 [Citation320]. Rodriguez et al. detected an expansion of eosinophils expressing CD62L (L-selectin) which is stimulated by IFN-γ and is widely expressed in COVID-19 so they suggested that the IFN-γ-eosinophil axis could contribute to hyperinflammation [Citation325].
2.8.4. Basophils
Basophils act at the interface between innate and adaptive immunity, they express Fc receptors for IgG and IgE so they have the ability to recognize and bind antigens and enhance the adaptive immune response, their degranulation releases several molecules including histamine, proteolytic enzymes and several cytokines mainly IL-4 and IL-13 that induce Th2 cell differentiation, in addition they can recruit and participate in inflammatory processes [Citation326]. The number of basophils decreases in critically ill patients compared to non-critically ill patients during SARS-CoV-2 infection and as recovery develops basophils show an increase in their levels [Citation44,Citation325,Citation327].
Dysregulation in T cells activity could also influence low basophil levels since it will decrease the production of T cell-derived cytokines such as IL-3 and GM-CSF that are associated with basophil production and survival [Citation328]. There is a correlation between basophil levels and the levels of IgG antibodies produced by B lymphocytes for SARS-CoV-2. Given that in COVID-19 originates a decrease in basophil levels, it has suggested that it could influence the efficacy of the IgG-mediated humoral immune response to SARS-CoV-2 [Citation325]. The role of basophils in COVID-19 and their influence in modulating the humoral immune response to SARS-CoV-2 remains to be further elucidated. Sun et al. reported that it is likely that individuals with a lower count or lower genetic predisposition for basophils might be more susceptible to severe COVID-19 [Citation329].
2.8.5. Natural killer (NK) cells
NK cells play an important role in viral infections, act as effector cells in the integration of innate and adaptive immunity, and also have memory characteristics that relate to the ability of these cells to recognize viruses directly [Citation330,Citation331]. They are rapidly recruited to the site of infection to eliminate and contain the virus, their activity is mediated by multiple receptors that respond to viral proteins, cellular stress molecules and antibody-coated cells by expressing Fc receptors, and they can also express receptors that trigger inhibitory signals [Citation331,Citation332]. NK cells could have a direct killing effect on SARS-CoV-2 infected cells through different mechanisms including cell apoptosis, release of molecules such as perforin and granzyme that can kill cells and by antibody-dependent cellular cytotoxicity as NK cells express CD16A that recognizes IgG antibodies bound to infected cells triggering the release of cytotoxic factors [Citation330,Citation333,Citation334].
In patients with COVID-19 the blood NK cell count decreases in relation to the severity status of the patient [Citation305,Citation335,Citation336]. But in deceased patients with COVID-19, Varchetta et al. detected that an increase of memory NK cells was manifested [Citation25]. The expression of cytotoxic molecules produced by NK cells was reduced in patients with severe COVID-19 relative to healthy patients [Citation25,Citation337]. During SARS-CoV-2 infection NK cells found in the circulation could be recruited to sites of infection such as the lungs, there NK cells would be active participating in inflammation and tissue damage [Citation331,Citation338,Citation339].
The reduction of NK cells favors SARS-CoV-2 in evading the immune response, several factors could be involved in their depletion and functional exhaustion. In COVID-19, NK cells increase the expression of NK cell receptor group 2, member A (NKG2A) which is related to functional depletion and inhibition of NK cell production [Citation339,Citation340]. Increased expression of mucin-domain- containing molecule-3 (TIM-3) and programmed cell death protein 1 (PD-1) which are molecules involved in functional depletion is recorded in NK cells, similarly reduced expression of NKG2D and IG-like lectin 7 which binds to sialic acid (Siglec-7) in COVID-19 which could alter NK cell activity as they are involved in potentiating their functionality [Citation25,Citation341]. Reduced levels of granzyme A-expressing NK cells are inversely correlated with IL-6 levels in blood of patients with COVID-19, which could indicate the involvement of IL-6 in impairing the cytotoxic activity of NK cells by negatively regulating granzyme A. expression [Citation337]. Bortolotti et al. observed that SARS-CoV-2 S1 protein might be able to modify the activation and effector mechanisms of NK cells as it can increase in the lungs the expression of HLA-E which is ligand of NKG2A thus inhibiting NK cell activity [Citation342]. NK cells have a high therapeutic potential for the treatment of COVID-19 there are even some clinical trials in development regarding COVID-19 [Citation333,Citation334].
2.8.6. Mast cells
They are located at various sites in the human body including skin, mucosa, respiratory tract, nasal cavity and intestine, participate in adaptive immune responses and are involved in responses against viral infections and inflammatory processes [Citation343,Citation344]. SARS-CoV-2 could induce mast cell activation through TLR or by induction of the high-affinity IgE receptor (FcεRI) that is expressed on mast cells and binds IgE [Citation343–345]. Mast cells produce molecules such as histamine, heparin, proteases, TNF-α, prostaglandin D2 (PGD2) and E2, leukotrienes B4 and C4 (LTB4 and LTC4), IL-1 β, IL-2, IL-4, IL-6, IL-18, TGF-β, CCL2, nerve growth factor (NGF) and vascular endothelial growth factor (VEGF), increases in several of these molecules are implicated in the pathogenesis of COVID. −19 [Citation345]. Expression of LTC4, PGD2 and histamine is related to increased mucus production and cough [Citation343].
SARS-CoV-2 can stimulate mast cells and increase the release of cytokines, chemokines and other molecules that could contribute to the formation of edema, inflammation and pulmonary fibrosis [Citation346]. Gebremeskel et al. provided evidence that active mast cells are associated with inflammation in COVID-19, due to elevated levels of specific mast cell-derived proteases and that TLR stimulation promotes their activation during COVID-19 [Citation347]. In post-mortem lung biopsies of COVID-19 detected an increase of mast cells mainly in the alveolar sacs, bronchioles terminals and alveolar septa, suggesting that mast cells may be involved in the pathogenesis of COVID-19 [Citation348].
2.8.7. Dendritic cells (DCs)
DC are potent APCs activate T lymphocytes through the major histocompatibility complex (MHC) [Citation349,Citation350]. There are two main groups of DC in blood: plasmacytoid dendritic cells (pDCs) and conventional dendritic cells (cDCs), each group of dendritic cells differ in their location and have several specific functions [Citation40,Citation351]. There is also a group of inflammatory dendritic cells characterized by the expression of carbohydrate 6-sulfo LacNAc (slanDCs) that produce high amounts of proinflammatory cytokines such as IL-12, IL-23, IL-1β and TNF- α [Citation352] DCs can generate the expression of different proinflammatory cytokines due to different stimuli that can activate various signaling pathways as they express different PRRs including TLR7, TLR8 and TLR9 that could recognize SARS-CoV-2 [Citation350]. Patients with COVID-19 showed decreased levels of pDCs, cDCs and slanDCs which could alter the immune response and contribute to the pathogenesis of the virus [Citation353–355]. Upon recognition and capture of a pathogen DCs mature and move to lymphoid organs and lymph nodes where they activate T cells through antigen presentation by MHC I and II, also influencing the expression of co-stimulatory molecules such as CD80/CD86 and cytokine expression [Citation351]. Antigens must be processed to be presented to the MHC, this processing can be for endogenous antigens which are degraded by the proteasome and their fragments loaded on MHC I activating CD8+ T lymphocytes, exogenous antigens are degraded in endosomes and their fragments are loaded on MHC II activating CD4+ T lymphocytes [Citation349,Citation351].
DCs in patients with COVID-19 show to have impaired T cell maturation and activation [Citation356]. Since the ability of DCs to present antigens is reduced during SARS-CoV-2 infection [Citation357]. The co-stimulatory proteins CD80 and CD86 that are important for T lymphocyte activation are similarly affected in COVID-19 [Citation356,Citation358]. Antigen presentation may also be altered by the development of apoptosis in DCs because SARS-CoV-2 could generate a state of metabolic stress and induce cell death [Citation359]. SARS-CoV-2 as a strategy to evade immune response alters the ability of DCs to produce IFN-I by inhibiting STAT1 phosphorylation [Citation298]. Onodi et al. showed that DCs are resistant to SARS-CoV-2 infection so they can express IFN-I and contribute with viral containment, the activation of DCs by SARS-CoV-2 depends on IRAK4 and UNC93B1 molecules in TLR signaling such as TLR 7 and 9 that are expressed in DCs [Citation360]. Monocyte-derived DCs (mDCs) can be infected by SARS-CoV-2 in an ACE2-dependent manner, they observed that upon infection there was ineffective replication and yet the virus induced alterations in mDC activity in addition to the expression of cytokines such as IL-6 and IL-1β but in lower proportion than in macrophages indicating the involvement of DCs in the pathogenesis of SARS-CoV-2 [Citation256,Citation361]. Similarly, DCs can also be infected [Citation362]. There is a positive up-regulation of Furin and DC-SING expression levels in DCs during SARS-CoV2 infection which could further contribute to virus entry [Citation298].
DCs are strong inducers of circulating soluble IL-6 receptor (sIL-6R) expression which can regulate free IL-6 in blood, DCs could contribute with the generation of an excess of sIL-6R over its inhibitor which is soluble glycoprotein 130 (sgp130), this imbalance allows trans IL-6 signaling to develop favoring an inflammatory state as in COVID-19 [Citation363]. IL-6 has two signaling pathways: a classical pathway that is generally anti-inflammatory and a trans pathway that is pro-inflammatory, the hyperinflammation in COVID-19 could depend on trans signaling, this signaling contributes to several cell types that would not respond to IL-6 under other conditions as they do not express IL-6R now do respond and induce signaling which could further contribute with cytokine expression [Citation253,Citation364]. DCs are one of the main cells that could be used as immunotherapy against COVID-19 with the aim of sensitizing the immune system, DCs could be used in vaccines but one of the limiting factors for now for cell therapy is the high cost [Citation365,Citation366].
2.9. Complement system (CS)
It is composed of different proteins that fulfill several defense functions against microorganisms in both innate and adaptive immunity, its deregulated activation is involved in inflammation, lung injury and vascular damage in COVID-19 [Citation367]. CS consists of three activation pathways: classical pathway, lectin pathway and alternative pathway, the goal of all three pathways is the formation of membrane attack complex (MAC or C5b-9) and the generation of several proteins such as C3a, C3b and C5a that have important functions in host defense [Citation368,Citation369]. MAC can alter the cell membrane by inducing the formation of holes leading to cell death; C3a and C5a are two potent anaphylatoxins capable of recruiting neutrophils and macrophages that contribute to inflammatory processes; C3b has the ability to opsonize infected cells and mark pathogens for elimination by phagocytosis [Citation370]. In lung epithelial cells SARS-CoV-2 can broadly stimulate activation of genes expressing CS components mainly through activation of IFN-JAK1/2-STAT1 and NF-kB signaling pathways [Citation371].
SARS-CoV-2 infected patients have the presence of biomarkers such as (C1q, MASP-2, C3, C3c, C3c, C3d, C4d and C5b-9) indicating strong activation of the alternative pathway and lectin pathway during infection [Citation372–375]. Ma and co-workers observed increased components of the alternative pathway during COVID-19 that were associated with coagulation proteins and endothelial damage [Citation373]. The SARS-CoV-2 protein S could directly activate the alternative pathway of CS on the cell surface [Citation376]. SARS-CoV-2 protein N interacts extensively with mannose-binding lectin-associated serine protease 2 (MASP2) and induces CS activation through the lectin pathway [Citation377,Citation378]. The classical pathway could be activated in late stages of COVID-19 by the production of antibodies against SARS-CoV-2 [Citation378].
CS is one of the most induced defense systems during SARS-CoV-2 infection generating increased expression of C3a and C3b that act in interaction with their receptors and drive inflammation [Citation316,Citation371]. Significantly increased levels of the C5b-9 complex together with increased C3a were strongly associated with the severity level of patients [Citation379,Citation380]. Increased plasma and BALF levels of soluble C5a similarly correlated with COVID-19 severity level [Citation381,Citation382]. C5a acts through its receptor C5aR1 to induce the recruitment and activation of immune cells such as monocytes and neutrophils by stimulating the production of proinflammatory cytokines [Citation381]. Overactivation of CS with elevated C3 are associated with an increased likelihood of severe COVID-19 and death [Citation379,Citation383].
CS also promotes the coagulation cascade by enhancing TF expression in different cell types during COVID-19 [Citation316,Citation370]. There is an interaction between CS and the coagulation cascade as MASP-2 can process prothrombin into thrombin which in turn can cleave C3 and C5, the products generated as C5a drive NET formation and TF expression in neutrophils which favor the expression of proteins involved in coagulation and inflammation resulting in a positive feedback loop [Citation370,Citation384]. In patients with COVID-19 an increase in plasma levels of von Willebrand factor (vWF) which plays a key role in coagulation processes was detected [Citation373,Citation382]. Its level was correlated with the level of C5b-9 in the course of COVID-19, C5b-9 could influence the release of vWF and lead to platelet aggregation inducing a prothrombotic state [Citation382,Citation385]. The complement system stimulates the NET-platelet-TF-thrombin axis which is associated with the increased immunothrombosis observed in several patients with severe COVID-19 [Citation386].
2.10. Adaptive immunity
Adaptive immunity has a very important feature which is the generation of long-lasting immunological memory where specialized cells and antibodies participate [Citation42]. In COVID-19 adaptive immunity can contribute in both host defense and disease development [Citation387,Citation388]. It is divided into two types of immunity: first, cell-mediated immunity mediated by the activity of T lymphocytes (CD4+ and CD8+); and second, humoral immunity mediated by B lymphocytes and their production of antibodies [Citation40]. The dysregulation of innate immune cell activity that occurs in COVID-19 could affect the correct development of the protective role of adaptive immunity [Citation389].
Effector and memory T lymphocytes play an important role in pathogen clearance and long-lasting immunological memory against possible reinfections, the various T lymphocytes phenotypes are altered during COVID-19 [Citation390]. Cytotoxic CD8+ T cells are responsible for eliminating pathogens through the production of proinflammatory cytokines, granzymes and perforins, CD4+ T cells differentiate according to different stimulatory agents into distinct T helper cells (Th) populations that have specific functions (Th1, Th2, Th17, T follicular helper cells (TFH) and regulatory T cells (Treg)) can regulate the activity of CD8+ T cells and B cells [Citation41,Citation391] (). Memory T lymphocytes are preserved for prolonged times can be divided into different subsets: central memory T cells (TCM) that maintain circulating memory, effector memory T cells (TEM) that guard non-lymphoid tissues and tissue-resident memory T cells (TRM) that have a rapid response at the site of reinfection [Citation392,Citation393].
Both T cells and B cells participate in the protection of the organism against SARS-CoV-2 infection by providing cellular and humoral immunity that interact to establish an effective response against the virus [Citation394,Citation395]. A common feature among most patients with COVID-19 is the development of lymphopenia which is the reduction in the amount of CD4+ and CD8+ T cells decreasing further in severe patients, usually the T lymphocytes count is usually normal in mild patients [Citation265,Citation335,Citation387,Citation396]. A low lymphocyte count correlates with poor control of SARS-CoV-2 dissemination and enhanced pathogenesis [Citation387]. summarizes the characteristics of adaptive immunity cells against SARS-CoV-2. Despite the decrease in CD4+ and CD8+ T cells, there are studies that indicate that the cells could present a high activation and effector capacity in severe COVID-19 which favors the production of cytokines, perforins and granzymes [Citation41,Citation257]. The dysfunctional activity of CD4+ and CD8+ T cells in patients with COVID-19 could be reflected in the increased expression of different molecular markers such as PD-1, TIM-3, lymphocyte activation gene 3 (LAG3), cytotoxic T lymphocyte antigen 4 (CTLA-4), CD69 and NKG2, these markers could be the consequence of functional exhaustion or high level of T cell activation [Citation25,Citation391,Citation396].
Table 2. Summary of the characteristics of the innate immune response to SARS-CoV-2.
2.10.1. CD8+ T lymphocytes
In most patients the decrease in T lymphocytes is more prominent for CD8+ T cells than in CD4+ T cells [Citation402,Citation403]. One factor that could have a negative impact on T-cell survival and proliferation is the excessive amount of proinflammatory cytokines in the circulation during severe COVID-19 [Citation41]. The increased apoptosis during COVID-19 which is associated with the severity of the patients, could be a contributing factor with lymphopenia as it affects CD4+ and CD8+ T cells and also the extensive T cells infiltration to the lungs [Citation359,Citation388,Citation404].
Within lymphopenia in COVID-19 the proportion of different T cell phenotypes is altered, there is a decrease in some groups of mainly virgin T cells and an increase in the proportion of different subsets of effector and memory T cells [Citation390]. Critically ill patients differ from mild patients not only in the reduced number of T cells but also in their decreased effector capacity, in BALF of mild patients there is a higher proportion of CD8+ TRM cells with good effector functions and highly expanded [Citation290,Citation405]. TRM cells can be found in different tissues including the lungs where they can trigger an effective response to reinfections, TRM cells are widely detected persisting in lung tissue samples from convalescent patients 10 months after SARS-CoV-2 infection indicating that recovered patients may have long-lasting protection against future infections [Citation404].
Specific CD8+ T cells and CD4+ T cells can recognize and induce strong reactivity to different SARS-CoV-2 epitopes [Citation406,Citation407]. Specific T cells can be detected early in disease by COVID-19 and increase over time [Citation395]. SARS-CoV-2 specific CD8+ T cells have a more effector phenotype produce several cytokines mainly granzyme B, perforins in combination with IFN-γ and TNF-α [Citation257,Citation406,Citation408]. Gangaev et al. observed that SARS-CoV2-specific CD8+ T cells in patients with severe and critical COVID-19 in the acute phase of the disease are found to be dysfunctional, their effector function is limited as their cytokine production is reduced, they also show an elevated level of activation and inhibitory receptors [Citation409]. Memory CD4+ T cells and CD8+ T cells are widely detected in patients with COVID-19 and they can persist for a prolonged time after the convalescence phase [Citation409,Citation410]. CD8+ T cells in blood of patients with COVID-19 and patients in convalescent phase show an increased TEM effector memory phenotype and also of the terminally differentiated effector memory T cells (TEMRA) [Citation408,Citation411,Citation412].
2.10.2. CD4+ T lymphocytes
Th1 and Th17 type CD4+ T cells detected in BALF from mild patients are expanded and have effector functions that are able to resolve the infection unlike in critically ill patients where these cells were significantly downregulated, there is a more prominent enrichment of T cells expressing depletion patterns and altered Th1 cells that in general contribute with exacerbation of inflammation [Citation290]. In addition the Th2 response is also dysregulated and could contribute to the severity of patients [Citation413,Citation414]. SARS-CoV-2 specific CD4+ T cells exhibit a predominant Th1 response expressing IFN-γ, TNF-α and IL-2 especially in critically ill patients although cytokines specific to Th2 and Th17 responses are also detected [Citation408,Citation411,Citation415]. A circulating TFH cell response is widely detected in the course of COVID-19, these cells have the ability to stimulate B lymphocytes for antibody production [Citation406,Citation415,Citation416]. However, an increase of TFH cells with cytotoxic characteristics was reported that may influence the reduced activity of adaptive immunity as they may be detrimental to B cells [Citation417]. Memory CD4+ T cells detected in blood from COVID-19 and convalescent patients presented in higher proportion a TCM memory phenotype [Citation411,Citation416,Citation418].
Memory CD4+ T and CD8+ T lymphocytes decrease over time, however, eight months after patients had COVID-19 they observed that most were still able to produce a cellular immune response against the virus [Citation419–421]. Lymphocytes are important cells in the production of proinflammatory cytokines, their decrease in patients with COVID-19 could indicate that they are not the main cause of the hyperinflammatory state but that there are alternative sources of the elevated cytokine production, however, although the number of lymphocytes is reduced, they were active and could therefore in part contribute to the pathogenesis of SARS-CoV-2 [Citation41]. The elicitation of memory T cells sometime after infection indicates that the organism can defend itself against possible reinfections of SARS-CoV-2 [Citation393,Citation422].
As for the level of regulatory T cells (Treg) is affected in severe COVID-19, these cells are in charge of maintaining homeostasis in uncontrolled immune responses, decreasing cytokine storm and reducing tissue damage so the decrease of their level could contribute with immune imbalance during the disease [Citation44,Citation403,Citation423]. Treg cells are associated with CD4+ and CD8+ T lymphocytes dysregulation so they might have some role in the pathogenesis of COVID-19 [Citation424].
2.10.3. B lymphocytes
In SARS-CoV-2 infection B cells can be activated and recognize epitopes directly or in a T lymphocytes-dependent manner differentiating into plasma cells that produce antibodies to eliminate pathogens and memory cells that remain for a secondary response against the same pathogen [Citation425]. The number of B cells decreases in COVID-19 in especially severely symptomatic and deceased patients [Citation426–428]. Although there are also studies in which B lymphocytes were found to be at normal levels and in some cases increased during COVID-19 [Citation403]. In severe patients, B cells show a deregulated activation that could favor a delayed humoral response contributing to the infectivity of the virus and the severity of the patients [Citation429]. The B lymphocytes response develops at least one week after symptom onset [Citation415]. Patients with COVID.19 generate IgA, IgM and IgG responses against SARS-CoV-2 proteins mainly specific for RBD, protein S and protein N [Citation394,Citation406,Citation430].
Serum antibody levels are related to the severity of the patients as in severe patients antibodies increase approximately 10 times than in mild patients [Citation395,Citation415,Citation431]. Neutralizing antibodies are important in protective immunity, they prevent viral particles from entering cells so these antibodies can be targeted and recognize various regions of the SARS-CoV-2 S protein to inhibit its infectivity by blocking viral entry [Citation430]. Antibodies targeting the RBD and NTD regions of the S protein was identified to show high potency to neutralize SARS-CoV-2 [Citation432,Citation433]. The response of IgA antibodies was evaluated in several patients in serum, saliva and BALF where they were found to dominate the early response to infection and contribute to virus neutralization, even at a higher level than IgG in the first weeks of the disease mainly at the mucosal level [Citation434].
Since SARS-CoV 2 enters through the upper respiratory tract, it is necessary to highlight the role of the protective immunity of the mucosa. On the mucosal surface, humoral immunity is mainly mediated by secretory IgA (sIgA) antibodies, which have an important role in the early defense of mucosal surfaces [Citation319,Citation425]. SIgA could help neutralize the entry of SARS-CoV-2 on the mucosa surface before it infects epithelial cells [Citation34]. The protective activity of sIgA has prompted the development of intranasal vaccines to improve mucosal immunity and thus limit the spread of SARS-CoV-2 [Citation34,Citation435].
Xu et al. reported a mean of 5.5 days to seroconversion of neutralizing antibodies directed to the RBD of SARS-CoV-2 after symptom onset [Citation436]. Orner et al. detected in patients with COVID-19 that seroconversion for IgG and IgM was 8 and 7 days respectively after symptom onset [Citation437]. Fu et al. found that the seroconversion rate peaked at 4–5 weeks [Citation438]. Ng et al. observed that seroconversion for IgG, IgM and neutralizing antibodies averaged 10.3 to 11 days [Citation439]. Lou et al. detected a mean of 9, 10 and 12 days after disease onset for total antibodies, IgM and IgG respectively [Citation440] In general antibodies specific for SARS-CoV-2 appear two weeks after symptom onset, the appearance of IgG and IgA occurs simultaneously or sequentially to the appearance of IgM [Citation441] (). Detection of IgM and IgA antibodies that bind to SARS-CoV-2 protein S are detected more frequently than IgG during the first two weeks after symptom onset but then decline steadily, while IgG remains elevated longer [Citation441,Citation443].
Figure 3. Specific antibody response in patients infected with COVID-19: The immune response is heterogeneous among patients; an overview of SARS-CoV-2 specific antibody formation is shown in this figure. The appearance of specific IgA, IgM and IgG antibodies usually occurs in the second week after symptom onset. IgG antibodies develop equal or in sequence to IgM antibodies and their levels are maintained for a longer time while IgM and IgA antibodies decline. The detection of protective antibodies with neutralizing activity may persist until at least 13 months after the onset of the disease. The virus incubation period on average can last 1-week Zaki and Mohamed obtained a mean of 7.8 days from the analysis of several studies [Citation442]. The amount of viable virus in individuals is variable usually decreases between 7-8 days from symptom onset but viral RNA can be detected for several weeks [Citation441]. Memory T and B cells have shown an increase and stability for at least 8 months from disease onset.
![Figure 3. Specific antibody response in patients infected with COVID-19: The immune response is heterogeneous among patients; an overview of SARS-CoV-2 specific antibody formation is shown in this figure. The appearance of specific IgA, IgM and IgG antibodies usually occurs in the second week after symptom onset. IgG antibodies develop equal or in sequence to IgM antibodies and their levels are maintained for a longer time while IgM and IgA antibodies decline. The detection of protective antibodies with neutralizing activity may persist until at least 13 months after the onset of the disease. The virus incubation period on average can last 1-week Zaki and Mohamed obtained a mean of 7.8 days from the analysis of several studies [Citation442]. The amount of viable virus in individuals is variable usually decreases between 7-8 days from symptom onset but viral RNA can be detected for several weeks [Citation441]. Memory T and B cells have shown an increase and stability for at least 8 months from disease onset.](/cms/asset/ec970a39-140d-4a53-be0e-f993d75b2d98/timm_a_2066251_f0003_c.jpg)
Persons recovered from COVID-19 may have a lower risk of reinfection with SARS-CoV-2 [Citation420]. The length of time that immunologic memory against SARS-CoV-2 lasts is still unknown but studies have evaluated at specific times. The number of SARS-CoV-2 protein-specific memory B cells increases several months after infection and then stabilizes for at least 8 months, which could indicate a long-lasting B cell memory [Citation420,Citation421,Citation444]. In addition, B cells evolve and produce antibodies that have increased resistance to RBD mutations, somatic hypermutation, potency and neutralization [Citation431]. Wisnivesky et al. evaluated long-term protective antibody titers and observed that most patients who had COVID-19 had persistent antibodies against SARS-CoV-2 protein S for up to 13 months after infection [Citation445]. Similarly, studies have detected neutralizing activity in recovered patients against virus proteins one year after symptom onset despite reduced antibody titers [Citation446,Citation447]. Other studies in a smaller number of patients detected that plasma neutralizing action as well as RBD-specific IgM and IgG antibody levels decrease significantly 6-7 months after infection and then gradually decline, but it is noteworthy that antibodies were detected at least 11 months after infection in several patients [Citation420,Citation431,Citation448]. The antibodies still detected may be the result of long-lived bone marrow plasma cells that are found in several patients recovered from COVID-19, these cells are a source of protective antibodies thus providing long-lasting immunity [Citation444]. In addition, another source of antibodies are memory B lymphocytes that are maintained over time and if exposed again to the same pathogen rapidly differentiate into protective antibody secreting cells [Citation448]. All the studies performed show an immunity that can be long-lasting in patients recovered by COVID-19 so it can provide protection against possible future reinfections [Citation444] ().
Table 3. Characteristics of the adaptive immune response to SARS-CoV-2 infection.
2.11. Immunopathology of COVID-19
2.11.1. Hyperinflammation
COVID-19 disease has multiple clinical presentations ranging from patients who generate no symptoms to patients who may have severe repercussions such as pneumonia, ARDS and death. One common feature in several patients with especially severe COVID-19 is an increase in proinflammatory cytokines and chemokines that can trigger a systemic hyperinflammatory state [Citation43,Citation257]. This state may be the consequence of SARS-CoV-2-induced dysregulation of the immune response [Citation291,Citation452]. During the initial stage of SARS-CoV-2 infection, the immune system could be immunodeficient favoring the spread of the virus, in the second instance the immune system generates a delayed hyperactive response to compensate for the failure of the early stage of the disease and try to eliminate the virus [Citation277]. In patients who died of COVID-19, Dorward et al. revealed that at several sites of inflammation in the lung there was no presence of viral RNA [Citation452]. This highlights the involvement of a hyperactive and aberrant SARS-CoV-2-induced immune response that may stimulate lung injury by inflammation [Citation452,Citation453].
The lung is the primary target of SARS-CoV-2 infection, in BALF from COVID-19 patients cytokines, chemokines and eicosanoids fuel hyperinflammation in the lung as well as elevated immune cell infiltration [Citation37]. Excessive release of inflammatory molecules can lead to the development of a cytokine storm that could be the trigger for multiple pathological manifestations in COVID-19 [Citation291,Citation454]. Elevated levels of inflammatory markers such as C-reactive protein and ferritin are found in patients with severe COVID-19 [Citation455,Citation456]. Lung cells sense SARS-CoV-2 through PRRs that stimulate different signaling pathways for the expression of several proinflammatory cytokines and chemokines mainly: IL-1β, IL-6, IL-8, GM-CSF, TNF-α, IFN-γ, IP-10, MCP-1, MIP-1α and MIP-1α [Citation291,Citation454]. A representative cytokine in COVID-19 is IL-6 its level increases and is associated with severe COVID-19 events so it is possible that it is one of the main contributors of the pulmonary hyperinflammatory state and organ damage just like IL-1 β and TNFα [Citation245,Citation246,Citation251].
Endothelial damage and cytokine release promotes the recruitment and activation of different immune cells such as macrophages, neutrophils and T lymphocytes that target the site of inflammation in the lungs where they can generate the expression of more cytokines and contribute to their accumulation [Citation308,Citation457]. Uncontrolled macrophage activity leading to MAS formation and uncontrolled neutrophil activity in producing proinflammatory cytokines is associated with the development of ARDS [Citation254]. One protein that may contribute extensively to cytokine storm is sterol element-binding protein-2 (SREBP-2) which regulates lipid and cholesterol biosynthesis [Citation458,Citation459]. SREBP-2 has an important role as an inflammatory transcription factor, it can regulate the production of IL-1β, TNF-α and IL-6, furthermore cholesterol biosynthesis by SREBP-2 is involved in the exocytosis (budding and envelopment) of SARS-CoV-2 from the cell [Citation458].
Several studies have taken cytokine storm as the predominant cause of severe disease in COVID-19 but there are discrepancies as Remy et al. have suggested that the main trigger for severity in COVID-19 is the failure or suppression of the immune response which allows the virus to spread easily causing organ damage [Citation460]. In COVID-19 an increased production of proinflammatory cytokines is observed but the concentrations are lower than those observed in non-COVID-19 associated ARDS, sepsis and cytokine release syndrome so there are suggestions that an inflammatory state induced by SARS-CoV-2 would not be the key determinant in the severity of the disease [Citation461]. Co-infections could be involved in the increased severity in patients with COVID-19 as they stimulate increased cytokine production [Citation254]. As well as people with an already pre-existing disease such as hypertension which can increase hyperinflammation in the airways of COVID-19 patients making them more susceptible to contracting severe episodes of the disease [Citation462]. Obesity is another disease that is associated with a poor prognosis of COVID-19 as it favors the progression of the hyperinflammatory state [Citation463]. Overall, the evidence provided by several studies suggests that hyperinflammation could be the main driver for the development of acute lung injury, severe pneumonia and ARDS which are the predominant cause of death in patients with COVID-19 [Citation453,Citation464].
2.11.2. Hypercoagulation
In several patients COVID-19 complications are associated with an altered coagulation status that directs the disease to its most severe or critical forms, even a large number of patients are at high risk for thrombotic complications [Citation465–467]. Several patients with COVID-19 have venous and arterial thrombotic events such as deep vein thrombosis [Citation468–470]. High prothrombotic state is observed in COVID-19 through significant increase in fibrinogen, dimer D, fibrin degradation product (FDP), factor VIII and vWF which are involved in coagulation processes, in addition abnormalities in platelet activity and platelet count are present [Citation456,Citation471,Citation472]. SARS-CoV-2 entry via ACE2 results in the accumulation of Ang II which is able to increase vasoconstriction and limit blood flow, direct infection of endothelial cells via ACE2 can induce elevated expression of plasminogen activators thus contributing to hypercoagulation [Citation471]. Platelets also contribute to thrombotic complications in patients with severe COVID-19 [Citation317,Citation473]. Stored vWF is released in COVID-19 and detected at elevated levels due to endothelial damage, this factor can interact with platelets and activate them promoting clot formation [Citation471,Citation474]. In COVID-19 an increase in platelet factor 4 (PF4) expression is detected which is released from platelets, PF4 binds to NET providing it with increased resistance and increases its expression [Citation307,Citation312].
Sustained inflammation promotes thrombosis formation, endothelial cell damage from excessive inflammation in COVID-19 leads to activation of coagulation and thrombus formation [Citation474–476]. SARS-CoV-2 and proinflammatory cytokines can stimulate TF expression in endothelial cells and monocytes promoting activation of the coagulation cascade [Citation477,Citation478]. Macrophage accumulation and MAS induction may influence the formation of pulmonary intravascular coagulation [Citation254]. The overactivated complement system in COVID-19 and NET may also be inducers of thrombotic problems as it can induce TF expression [Citation479,Citation480]. Several organs are affected by SARS-CoV-2-induced hypercoagulation, coagulation disorder can generate different pathologies such as increased risk of stroke and neuroinflammation [Citation343,Citation481]. Hypercoagulation may also be a contributing factor with acute kidney injury, cardiovascular problems that occur in patients with severe COVID-19 and pulmonary coagulation that may trigger the development of pneumonia and ARDS [Citation482,Citation483]. In we recapitulate a behavior of the protective immune system capable of limiting the spread of SARS-CoV-2 and the imbalance of the immune response induced by the virus generating a dysregulated immune response that influences the severity of patients through the development of immunopathological disorders.
Figure 4. Balance between protective immunity and the consequences of dysregulated immunity during SARS-CoV-2 infection.
3. Conclusion
SARS-CoV-2 has demonstrated special characteristics that allow it to enhance its infectivity and transmission, infecting millions of people worldwide. The immune system in general is a very complex defense mechanism, and its development in the face of SARS-CoV-2 can vary among each patient, resulting in different disease states. Since the inception of COVID-19 an enormous number of studies have endeavored to elucidate the mechanism behind the interaction of SARS-CoV-2 and the immune system to determine a better way to prevent infection, treat patients infected and possible reinfections. Several unknowns and controversies that have arisen from the various COVID-19 studies remain to be clarified.
Here we have recapitulated and analyzed a number of scientific papers to provide a generalized understanding of the characteristics of SARS-CoV-2 and also its interaction with each component of the immune system. The virus causes an initial response that is immunodeficient allowing it to infect cells with ease and cause different damages in the organism, then the immune system in order to compensate for the initial failure initiates a delayed hyperactive response which can lead to the different clinical features observed in patients. Therefore, the virus directly or indirectly through an aberrant immune response causes significant damage associated with inflammation and coagulation pathologies in infected patients. The majority of patients through the immune response can resolve the SARS-CoV-2 infection but there is another smaller percentage where the immune response plays a double-edged role in driving the severity of the patient which can lead to the fatal outcome which is death. In general, SARS-CoV-2 impairs the proper protective functioning of the immune system, inducing an innate immune response, complement system and adaptive immune response with overactivation characteristics. Information was obtained that may be encouraging since the memory T and B cells produced in recovered patients could generate a long-lasting immune response, so far it is evaluated for several months but it could last for years and would help to develop a better immune defense against possible reinfections. The review also managed to highlight the progress of COVID-19 immunopathology and attractive features that several studies consider to be therapeutic targets for the treatment of COVID-19.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Corman V, Muth D, Niemeyer D, et al. Hosts and sources of endemic human coronaviruses. Adv Virus Res. 2018;100:163–188.
- Haake C, Cook S, Pusterla N, et al. Coronavirus infections in companion animals: virology, epidemiology, clinical and pathologic features. Viruses. 2020;12(9):1023.
- Graham RL, Baric RS. Recombination, reservoirs, and the modular spike: mechanisms of coronavirus cross-species transmission. J Virol. 2010;84(7):3134–3146.
- Zhu N, Zhang D, Wang W, China Novel Coronavirus Investigating and Research Team, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382(8):727–733.
- Gorbalenya AE, Baker SC, Baric RS, et al. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5:536–544.
- Zhou P, Yang X, Lou Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273.
- Chen Z, Boon SS, Wang MH, et al. Genomic and evolutionary comparison between SARS-CoV-2 and other human coronaviruses. J Virol Methods. 2021;289(114032):114032.
- Zhang T, Wu Q, Zhang Z. Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr Biol. 2020;30(7):1346–1351.
- Zhu Z, Lian X, Su X, et al. From SARS and MERS to COVID-19: a brief summary and comparison of severe acute respiratory infections caused by three highly pathogenic human coronaviruses. Respir Res. 2020;21(1):224.
- Hasöksüz M, Kiliç S, Saraç F. Coronaviruses and SARS-COV-2. Turk J Med Sci. 2020;50(SI-1):549–556.
- Su S, Wong G, Shi W, et al. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016;24(6):490–502.
- Sariol A, Perlman S. Lessons for COVID-19 immunity from other coronavirus infections. Immunity. 2020;53(2):248–263.
- Poyraz BÇ, Poyraz CA, Olgun Y, et al. Psychiatric morbidity and protracted symptoms after COVID-19. Psychiatry Res. 2021;295(113604):113604.
- Iser BPM, Sliva I, Raymundo VT, et al. Suspected COVID-19 case definition: a narrative review of the most frequent signs and symptoms among confirmed cases. Epidemiol e Serv Saude. 2020;29(3):e2020233.
- Galbadage T, Peterson BM, Gunasekera RS. Does COVID-19 spread through droplets alone? Front Public Heal. 2020;8:163.
- Morawska L, Cao J. Airborne transmission of SARS-CoV-2: the world should face the reality. Environ Int. 2020;139:105730.
- Nazir R, Ali J, Rasul I, et al. Eco-environmental aspects of covid-19 pandemic and potential control strategies. IJERPH. 2021;18(7):3488.
- Rabaan AA, Al-Ahmed SH, Al-Malkey MK, et al. Airborne transmission of SARS-CoV-2 is the dominant route of transmission: droplets and aerosols. Infez Med. 2021;29(1):10–19.
- Belosi F, Conte M, Gianelle V, et al. On the concentration of SARS-CoV-2 in outdoor air and the interaction with pre-existing atmospheric particles. Environ Res. 2021;193(110603):110603.
- Wang Y, Xu G, Huang YW. Modeling the load of SARS-CoV-2 virus in human expelled particles during coughing and speaking. PLOS One. 2020;15(10):e0241539.
- Gu J, Han B, Wang J. COVID-19: gastrointestinal manifestations and potential fecal-oral transmission. Gastroenterology. 2020;158(6):1518–1519.
- Barrera-Nunez D, Torres-Ibarra L, Leon-Maldonado L, et al. L. Revisión rápida de la transmisión del SARS-CoV-2 por contacto con objetos y superficies. Salud Publica Mex. 2020;63(1, ene-feb):126–135.
- Marquès M, Domingo JL. Contamination of inert surfaces by SARS-CoV-2: persistence, stability and infectivity. A review. Environ Res. 2021;193(110559):110559.
- Dargahi A, Jeddi F, Vosoughi M, et al. Investigation of SARS CoV-2 virus in environmental surface. Environ Res. 2021;195(110765):110765.
- Varchetta S, Mele D, Oliviero B, et al. Unique immunological profile in patients with COVID-19. Cell Mol Immunol. 2021;18(3):604–612.
- Meyerowitz EA, Richterman A, Gandhi RT, et al. Transmission of SARS-CoV-2: a review of viral, host, and environmental factors. Ann Intern Med. 2021;174(1):69–79.
- Mondelli MU, Colaneri M, Seminari EM, et al. Low risk of SARS-CoV-2 transmission by fomites in real-life conditions. Lancet Infect Dis. 2021;21(5):e112.
- Ong SWX, Lee PH, Tan YK, et al. Environmental contamination in a COVID-19 intensive care unit (ICU) – what is the risk? Infect Control Hosp Epidemiol. 2021;42(6):669–677.
- Colaneri M, Seminari E, Novati S, et al. Severe acute respiratory syndrome coronavirus 2 RNA contamination of inanimate surfaces and virus viability in a health care emergency unit. Clin Microbiol Infect. 2020;26(8):1094.e1–1094–e5.
- Hirose R, Ikegaya H, Naito Y, et al. Survival of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and influenza virus on human skin: Importance of hand hygiene in coronavirus disease 2019 (COVID-19). Clin Infect Dis. 2021;73(11):e4329–e4335.
- Shah VK, Firmal P, Alam A, et al. Overview of immune response during SARS-CoV-2 infection: lessons from the past. Front Immunol. 2020;11:1949.
- Sungnak W, Huang N, Bécavin C, HCA Lung Biological Network, et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med. 2020;26(5):681–687.
- Hekman RM, Hume AJ, Goel RK, et al. Actionable cytopathogenic host responses of human alveolar type 2 cells to SARS-CoV-2. Mol Cell. 2020;80(6):1104–1122.e9.
- Lapuente D, Fuchs J, Willar J, et al. Protective mucosal immunity against SARS-CoV-2 after heterologous systemic prime-mucosal boost immunization. Nat Commun. 2021;12(1):6871.
- Sallenave JM, Guillot L. Innate immune signaling and proteolytic pathways in the resolution or exacerbation of SARS-CoV-2 in covid-19: key therapeutic targets? Front Immunol. 2020;11:1229.
- Ricci D, Etna MP, Rizzo F, et al. Innate immune response to sars‐cov‐2 infection: from cells to soluble mediators. IJMS. 2021;22(13):7017.
- Zaid Y, Doré É, Dubuc I, et al. Chemokines and eicosanoids fuel the hyperinflammation within the lungs of patients with severe COVID-19. J Allergy Clin Immunol. 148(2):368–380.e3.
- Khalil BA, Elemam NM, Maghazachi AA. Chemokines and chemokine receptors during COVID-19 infection. Comput Struct Biotechnol J. 2021;19:976–988.
- Trouillet-Assant S, Viel S, Gaymard A, COVID HCL Study group, et al. Type I IFN immunoprofiling in COVID-19 patients. J Allergy Clin Immunol. 2020;146(1):206–208.e2.
- Borges RC, Hohmann MS, Borghi SM. Dendritic cells in COVID-19 immunopathogenesis: insights for a possible role in determining disease outcome. Int Rev Immunol. 2021;40(1-2):108–125.
- de Candia P, Prattichizzo F, Garavelli S, et al. T cells: warriors of SARS-CoV-2 infection. Trends Immunol. 2021;42(1):18–30.
- Netea MG, Schlitzer A, Placek K, et al. Innate and adaptive immune memory: an evolutionary continuum in the host’s response to pathogens. Cell Host Microbe. 2019;25(1):13–26.
- Hosseini A, Hashemi V, Shomali N, et al. Innate and adaptive immune responses against coronavirus. Biomed Pharmacother. 2020;132(110859):110859.
- Qin C, Zhou L, Hu Z, et al. Dysregulation of immune response in patients with Coronavirus 2019 (COVID-19) in Wuhan, China. Clin Infect Dis. 2020;71(15):762–768.
- Jose RJ, Manuel A. COVID-19 cytokine storm: the interplay between inflammation and coagulation. Lancet Respir Med. 2020;8(6):e46–e47.
- Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506.
- Park A, Iwasaki A. Type I and type III interferons – induction, signaling, evasion, and application to combat COVID-19. Cell Host Microbe. 2020;27(6):870–878.
- Li G, Fan Y, Lai Y, et al. Coronavirus infections and immune responses. J Med Virol. 2020;92(4):424–432.
- Shi Y, Wang Y, Shao C, et al. COVID-19 infection: the perspectives on immune responses. Cell Death Differ. 2020;27(5):1451–1454.
- Root-Bernstein R. Innate receptor activation patterns involving tlr and nlr synergisms in COVID-19, ALI/ARDS and sepsis cytokine storms: a review and model making novel predictions and therapeutic suggestions. IJMS. 2021;22(4):2108.
- Coperchini F, Chiovato L, Croce L, et al. The cytokine storm in COVID-19: an overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 2020;53:25–32.
- Seyed Hosseini E, Kashani NR, Nikzad H, et al. The novel coronavirus disease-2019 (COVID-19): mechanism of action, detection and recent therapeutic strategies. Virology. 2020;551:1–9.
- Li X, Geng M, Peng Y, et al. Molecular immune pathogenesis and diagnosis of COVID-19. J Pharm Anal. 2020;10(2):102–108.
- Chan JFW, Kok KH, Zhu Z, et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. 2020;9(1):221–236.
- Rohaim MA, El Naggar RF, Clayton E, et al. Structural and functional insights into non-structural proteins of coronaviruses. Microb Pathog. 2021;150(104641):104641.
- Mousavizadeh L, Ghasemi S. Genotype and phenotype of COVID-19: their roles in pathogenesis. J Microbiol Immunol Infect. 2021;54(2):159–163.
- Amanat F, Krammer F. SARS-CoV-2 vaccines: status report. Immunity. 2020;52(4):583–589.
- Tian X, Li C, Huang A, et al. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg Microbes Infect. 2020;9(1):382–385.
- Sternberg A, Naujokat C. Structural features of coronavirus SARS-CoV-2 spike protein: targets for vaccination. Life Sci. 2020;257:118056.
- Walls AC, Tortorici MA, Bosch BJ, et al. Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature. 2016;531(7592):114–117.
- Walls AC, Park YJ, Tortorici MA, et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181(2):281–292.
- Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280.
- Scialo F, Daniele A, Amato F, et al. ACE2: the major cell entry receptor for SARS-CoV-2. Lung. 2020;198(6):867–877.
- Santos RAS, Sampaio WO, Alzamora AC, et al. The ACE2/angiotensin-(1-7)/MAS axis of the renin-angiotensin system: focus on angiotensin-(1-7). Physiol Rev. 2018;98(1):505–553.
- Mirabito KM, Bovée DM, Danser AHJ. The renin-angiotensin-aldosterone system and its therapeutic targets. Exp Eye Res. 2019;186:107680.
- Datta PK, Liu F, Fischer T, et al. SARS-CoV-2 pandemic and research gaps: Understanding SARS-CoV-2 interaction with the ACE2 receptor and implications for therapy. Theranostics. 2020;10(16):7448–7464.
- Danser AHJ, Epstein M, Batlle D. Renin-angiotensin system blockers and the COVID-19 pandemic: at present there is no evidence to abandon renin-angiotensin system blockers. Hypertension. 2020;75(6):1382–1385.
- Mahmudpour M, Roozbeh J, Keshavarz M, et al. COVID-19 cytokine storm: the anger of inflammation. Cytokine. 2020;133(155151):155151.
- Peiró C, Moncada S. Substituting angiotensin-(1-7) to prevent lung damage in SARS-CoV-2 infection? Circulation. 2020;141(21):1665–1666.
- Bitker L, Burrell LM. Classic and nonclassic renin-angiotensin systems in the critically ill. Crit Care Clin. 2019;35(2):213–227.
- Kai H, Kai M. Interactions of coronaviruses with ACE2, angiotensin II, and RAS inhibitors-lessons from available evidence and insights into COVID-19. Hypertens Res. 2020;43(7):648–654.
- Zolfaghari Emameh R, Falak R, Bahreini E. Application of system biology to explore the association of neprilysin, angiotensin-converting enzyme 2 (ACE2), and carbonic anhydrase (CA) in pathogenesis of SARS-CoV-2. Biol Proced Online. 2020;22:11.
- El Tabaa MM, El Tabaa MM. Targeting neprilysin (NEP) pathways: a potential new hope to defeat COVID-19 ghost. Biochem Pharmacol. 2020;178:114057.
- Bayes-Genis A, Barallat J, Richards AM. A test in context: Neprilysin: function, inhibition, and biomarker. J Am Coll Cardiol. 2016;68(6):639–653.
- Campbell DJ. Neprilysin inhibitors and bradykinin. Front Med. 2018;5:257.
- Esser N, Zraika S. Neprilysin inhibitors and angiotensin(1-7) in COVID-19 nathalie. Br J Cardiol. 2020;27(4):109–111.
- Bellis A, Mauro C, Barbato E, et al. The rationale for angiotensin receptor neprilysin inhibitors in a multi-targeted therapeutic approach to COVID-19. IJMS. 2020;21(22):8612.
- Mehrabadi ME, Hemmati R, Tashakor A, et al. Induced dysregulation of ACE2 by SARS-CoV-2 plays a key role in COVID-19 severity. Biomed Pharmacother. 2021;137(111363):111363.
- Zhang L, Zetter MA, Guerra EC, et al. ACE2 in the second act of COVID-19 syndrome: peptide dysregulation and possible correction with oestrogen. J Neuroendocrinol. 2021;33(2):e12935.
- Liu Y, Yang Y, Zhang C, et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. 2020;63(3):364–374.
- Baig AM, Khaleeq A, Ali U, et al. Evidence of the COVID-19 virus targeting the CNS: tissue distribution, host-virus interaction, and proposed neurotropic mechanisms. ACS Chem Neurosci. 2020;11(7):995–998.
- Bian J, Li Z. Angiotensin-converting enzyme 2 (ACE2): SARS-CoV-2 receptor and RAS modulator. Acta Pharm Sin B. 2021;11(1):1–12.
- Ni W, Yang X, Yang D, et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit Care. 2020;24(1):422.
- Lee I-C, Huo T-I, Huang Y-H. Gastrointestinal and liver manifestations in patients with COVID-19. J Chinese Med Assoc. 2020;83(6):521–523.
- Sardu C, Gambardella J, Morelli MB, et al. Hypertension, thrombosis, kidney failure, and diabetes: is COVID-19 an endothelial disease? A comprehensive evaluation of clinical and basic evidence. JCM. 2020;9(5):1417.
- Shang J, Wan Y, Luo C, et al. Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci USA. 2020;117(21):11727–11734.
- Chen Y, Guo Y, Pan Y, et al. Structure analysis of the receptor binding of 2019-nCoV. Biochem Biophys Res Commun. 2020;525(1):135–140.
- Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol. 2020;5(4):562–569.
- Wrapp D, Wang N, Corbett KS, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–1263.
- Lan J, Ge J, Yu J, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215–220.
- Benton DJ, Wrobel AG, Xu P, et al. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature. 2020;588(7837):327–330.
- Cai Y, Zhang J, Xiao T, et al. Distinct conformational states of SARS-CoV-2 spike protein. Science. 2020;369(6511):1586–1592.
- Kirtipal N, Bharadwaj S, Gu S. From SARS to SARS-CoV-2, insights on structure, pathogenicity and immunity aspects of pandemic human coronaviruses. Infect Genet Evol. 2020;85:104502.
- V’kovski P, Kratzel A, Steiner S, et al. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol. 2021;19(3):155–170.
- Clausen TM, Sandoval DR, Spliid CB, et al. SARS-CoV-2 infection depends on cellular heparan sulfate and ACE2. Cell. 2020;183(4):1043–1057.e15.
- Chu H, Hu B, Huang X, et al. Host and viral determinants for efficient SARS-CoV-2 infection of the human lung. Nat Commun. 2021;12(1):134.
- Yu M, Zhang T, Zhang W, et al. Elucidating the interactions between heparin/heparan sulfate and SARS-CoV-2-related proteins-an important strategy for developing novel therapeutics for the COVID-19 pandemic. Front Mol Biosci. 2020;7:628551.
- Dakal TC. SARS-CoV-2 attachment to host cells is possibly mediated via RGD-integrin interaction in a calcium-dependent manner and suggests pulmonary EDTA chelation therapy as a novel treatment for COVID 19. Immunobiology. 2021;226(1):152021.
- Carvacho I, Piesche M. RGD-binding integrins and TGF-β in SARS-CoV-2 infections – novel targets to treat COVID-19 patients? Clin Transl Immunol. 2021;10(3):e1240.
- Park EJ, Myint PK, Appiah MG, et al. The spike glycoprotein of SARS-CoV-2 binds to β1 integrins expressed on the surface of lung epithelial cells. Viruses. 2021;13(4):645.
- Ludwig BS, Kessler H, Kossatz S, et al. Rgd-binding integrins revisited: How recently discovered functions and novel synthetic ligands (re-)shape an ever-evolving field. Cancers. 2021;13(7):1711.
- Su H, Wan C, Wang Z-D, et al. Expression of CD147 and cyclophilin a in kidneys of patients with COVID-19. Clin J Am Soc Nephrol. 2021;16(4):618–619.
- Wang K, Chen W, Zhang Z, et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct Target Ther. 2020;5:283.
- Radzikowska U, Ding M, Tan G, et al. Distribution of ACE2, CD147, CD26, and other SARS-CoV-2 associated molecules in tissues and immune cells in health and in asthma, COPD, obesity, hypertension, and COVID-19 risk factors. Allergy. 2020;75(11):2829–2845.
- Guindolet D, Gabison EE. Role of CD147 (EMMPRIN/basigin) in tissue remodeling. Anat Rec. 2020;303(6):1584–1589.
- Xin X, Zeng X, Gu H, et al. CD147/EMMPRIN overexpression and prognosis in cancer: a systematic review and meta-analysis. Sci Rep. 2016;6:32804.
- Rahimi N. C-type lectin CD209L/L-SIGN and CD209/DC-SIGN: Cell adhesion molecules turned to pathogen recognition receptors. Biology. 2020;10(1):1.
- Brufsky A, Lotze MT. DC/L-SIGNs of hope in the COVID-19 pandemic. J Med Virol. 2020;92(9):1396–1398.
- Bestle D, Heindl MR, Limburg H, et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci Alliance. 2020;3(9):e202000786.
- Tang T, Jaimes JA, Bidon MK, et al. Proteolytic activation of SARS-CoV-2 spike at the S1/S2 boundary: potential role of proteases beyond furin. ACS Infect Dis. 2021;7(2):264–272.
- Jackson CB, Farzan M, Chen B, et al. Mechanisms of SARS-CoV-2 entry into cells. Nat Rev Mol Cell Biol. 2022;23(1):3–20.
- Sarker J, Das P, Sarker S, et al. A review on expression, pathological roles, and inhibition of TMPRSS2, the serine protease responsible for SARS-CoV-2 spike protein activation. Scientifica. 2021;2021:2706789.
- Fuentes-Prior P. Priming of SARS-CoV-2 S protein by several membrane-bound serine proteinases could explain enhanced viral infectivity and systemic COVID-19 infection. J Biol Chem. 2021;296:100135.
- Katopodis P, Anikin V, Randeva HS, et al. Pan-cancer analysis of transmembrane protease serine 2 and cathepsin L that mediate cellular SARS.CoV.2 infection leading to COVID-19. Int J Oncol. 2020;57(2):533–539.
- Hoffmann M, Kleine-Weber H, Pöhlmann S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol Cell. 2020;78(4):779–784.e5.
- Drak Alsibai K. Expression of angiotensin-converting enzyme 2 and proteases in COVID-19 patients: a potential role of cellular FURIN in the pathogenesis of SARS-CoV-2. Med Hypotheses. 2020;143:109893.
- Martinez Z, Grönholm A, Kytölä V, et al. Myeloid cell expressed proprotein convertase FURIN attenuates inflammation. Oncotarget. 2016;7(34):54392–54404.
- Braun E, Sauter D. Furin-mediated protein processing in infectious diseases and cancer. Clin Transl Immunol. 2019;8(8):e1073.
- Fernandez C, Rysä J, Almgren P, et al. Plasma levels of the proprotein convertase furin and incidence of diabetes and mortality. J Intern Med. 2018;284(4):377–387.
- Swärd P, Rosengren BE, Jehpsson L, et al. Association between circulating furin levels, obesity and pro-inflammatory markers in children. Acta Paediatr. 2021;110(6):1863–1868.
- AbdelMassih AF, Ye J, Kamel A, et al. A multicenter consensus: a role of furin in the endothelial tropism in obese patients with COVID-19 infection. Obes Med. 2020;19:100281.
- Jin X, Xu K, Jiang P, et al. Virus strain of a mild COVID-19 patient in hangzhou represents a new trend in SARS-CoV-2 evolution related to furin cleavage site. Emerg Microbes Infect. 2020;9(1):1474–1488.
- Zhao MM, Yang WL, Yang FY. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct Target Ther. 2021;6:134.
- Gomes CP, Fernandes DE, Casimiro F, et al. Cathepsin L in COVID-19: from pharmacological evidences to. Genetics. Front Cell Infect Microbiol. 2020;10:589505.
- Padmanabhan P, Desikan R, Dixit NM. Targeting TMPRSS2 and cathepsin B/L together may be synergistic against SARSCoV- 2 infection. PLoS Comput Biol. 2020;16(12):e1008461.
- Qian Z, Dominguez SR, Holmes KV. Role of the spike glycoprotein of human Middle east respiratory syndrome coronavirus (MERS-CoV) in virus entry and syncytia formation. PLOS One. 2013;8(10):e76469.
- Bertram S, Glowacka I, Blazejewska P, et al. TMPRSS2 and TMPRSS4 facilitate trypsin-independent spread of influenza virus in caco-2 cells. J Virol. 2010;84(19):10016–10025.
- Zang R, Castro MFG, McCune BT, et al. TMPRSS2 and TMPRSS4 mediate SARS-CoV-2 infection of human small intestinal enterocytes. Sci Immunol. 2020;5(47):eabc3582.
- Villalba M, Redin E, Exposito F, et al. Identification of a novel synthetic lethal vulnerability in non-small cell lung cancer by co-targeting TMPRSS4 and DDR1. Sci Rep. 2019;9(1):15400.
- Chikaishi Y, Uramoto H, Koyanagi Y, et al. TMPRSS4 expression as a marker of recurrence in patients with lung cancer. Anticancer Res. 2016;36(1):121–127.
- Nguyen TH, Weber W, Havari E, et al. Expression of TMPRSS4 in non-small cell lung cancer and its modulation by hypoxia. Int J Oncol. 2012;41(3):829–838.
- Villalba M, Diaz-Lagares A, Redrado M, et al. Epigenetic alterations leading to TMPRSS4 promoter hypomethylation and protein overexpression predict poor prognosis in squamous lung cancer patients. Oncotarget. 2016;7(16):22752–22769.
- Han G, Sinjab A, Hara K, et al. Single-cell expression landscape of SARS-CoV-2 receptor ACE2 and host proteases in normal and malignant lung tissues from pulmonary adenocarcinoma patients. Cancers. 2021;13(6):1250.
- Dai M, Liu D, Liu M, et al. Patients with cancer appear more vulnerable to SARS-CoV-2: a multicenter study during the COVID-19 outbreak. Cancer Discov. 2020;10(6):783–791.
- Liang W, Guan W, Chen R, et al. Cancer patients in SARS-CoV-2 infection: a nationwide analysis in China. Lancet Oncol. 2020;21(3):335–337.
- Addeo A, Friedlaender A. Cancer and COVID-19: unmasking their ties. Cancer Treat Rev. 2020;88:102041.
- Katopodis P, Kerslake R, Davies J, et al. COVID-19 and SARS-CoV-2 host cell entry mediators: expression profiling of TMRSS4 in health and disease. Int J Mol Med. 2021;47(4):64.
- Hoffmann M, Hofmann-Winkler H, Smith JC, et al. Camostat mesylate inhibits SARS-CoV-2 activation by TMPRSS2-related proteases and its metabolite GBPA exerts antiviral activity. EBioMedicine. 2021;65:103255.
- Wruck W, Adjaye J. SARS-CoV-2 receptor ACE2 is co-expressed with genes related to transmembrane serine proteases, viral entry, immunity and cellular stress. Sci Rep. 2020;10(1):21415.
- Kishimoto M, Uemura K, Sanaki T, et al. TMPRSS11D and TMPRSS13 activate the SARS-CoV-2 spike protein. Viruses. 2021;13(3):384.
- Zhang C, Zhang Y, Zhang S, et al. Intracellular autoactivation of TMPRSS11A, an airway epithelial transmembrane serine protease. J Biol Chem. 2020;295(36):12686–12696.
- Cantuti-Castelvetri L, Ojha R, Pedro LD, et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. 2020;370(6518):856–860.
- Daly JL, Simonetti B, Antón-Plágaro C, et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science. 2020;370(6518):861–865.
- McFarland AJ, Yousuf MS, Shiers S, et al. Neurobiology of SARS-CoV-2 interactions with the peripheral nervous system: implications for COVID-19 and pain. Pain Rep. 2021;6(1):e885.
- Li Z L, Buck M. Neuropilin-1 assists SARS-CoV-2 infection by stimulating the separation of spike protein S1 and S2. Biophys J. 2021;120(14):2828–2837.
- Jobe A, Vijayan R. Neuropilins: C-end rule peptides and their association with nociception and COVID-19. Comput Struct Biotechnol J. 2021;19:1889–1895.
- Alnomasy SF. Virus-receptor interactions of SARS-CoV-2 spikereceptor-binding domain and human neuropilin-1 b1 domain. Saudi J Biol Sci. 2021;28(7):3926–3928.
- Hopkins C, Lechien JR, Saussez S. More that ACE2? NRP1 may play a Central role in the underlying pathophysiological mechanism of olfactory dysfunction in COVID-19 and its association with enhanced survival. Med Hypotheses. 2021;146(110406):110406.
- Mayi BS, Leibowitz JA, Woods AT, et al. The role of Neuropilin-1 in COVID-19. PLoS Pathog. 2021;17(1):e1009153.
- Liu C, Somasundaram A, Manne S, et al. Neuropilin-1 is a T cell memory checkpoint limiting long-term antitumor immunity. Nat Immunol. 2020;21(9):1010–1021.
- Vique-Sánchez JL. Potential inhibitors interacting in neuropilin-1 to develop an adjuvant drug against COVID-19, by molecular docking. Bioorganic Med Chem. 2021;33(116040):116040.
- Perez-Miller S, Patek M, Moutal A, et al. Novel compounds targeting neuropilin receptor 1 with potential to interfere with SARS-CoV-2 virus entry. ACS Chem Neurosci. 2021;12(8):1299–1312.
- Zunke F, Rose-John S. The shedding protease ADAM17: physiology and pathophysiology. Biochim Biophys Acta Mol Cell Res. 2017;1864(11 Pt B):2059–2070.
- Gooz M. ADAM-17: the enzyme that does it all. Crit Rev Biochem Mol Biol. 2010;45(2):146–169.
- Schumacher N, Rose-John S. ADAM17 activity and IL-6 Trans-Signaling in inflammation and cancer. Cancers. 2019;11(11):1736.
- Gheblawi M, Wang K, Viveiros A, et al. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circ Res. 2020;126(10):1456–1475.
- Heurich A, Hofmann-Winkler H, Gierer S, et al. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol. 2014;88(2):1293–1307.
- Pérez-Vizcaíno F. Lung ACE2 and ADAM17 in pulmonary arterial hypertension: implications for COVID-19? J Hear Lung Transplant. 2020;39(10):1167–1169.
- Monteil V, Kwon H, Prado P, et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. 2020;181(4):905–913.
- Sharma RK, Li J, Krishnan S, et al. Angiotensin-converting enzyme 2 and COVID-19 in cardiorenal diseases. Clin Sci. 2021;135(1):1–17.
- Xu J, Sriramula S, Xia H, et al. Clinical relevance and role of neuronal AT1 receptors in ADAM17-mediated ACE2 shedding in neurogenic hypertension. Circ Res. 2017;121(1):43–55.
- Zipeto D, Palmeira J, da F, et al. ACE2/ADAM17/TMPRSS2 interplay may be the main risk factor for COVID-19. Front Immunol. 2020;11:576745.
- Hall KC, Blobel CP. Interleukin-1 stimulates ADAM17 through a mechanism independent of its cytoplasmic domain or phosphorylation at threonine 735. PLOS One. 2012;7(2):e31600.
- Garvin MR, Alvarez C, Miller JI, et al. A mechanistic model and therapeutic interventions for covid-19 involving a ras-mediated bradykinin storm. Elife. 2020;9:e59177.
- van de Veerdonk FL, Netea MG, van Deuren M, et al. Kallikrein-kinin blockade in patients with covid-19 to prevent acute respiratory distress syndrome. Elife. 2020;9:e57555.
- Roche JA, Roche R. A hypothesized role for dysregulated bradykinin signaling in COVID-19 respiratory complications. Faseb J. 2020;34(6):7265–7269.
- Wilczynski SA, Wenceslau CF, Mccarthy CG, et al. A cytokine/bradykinin storm comparison: what is the relationship between hypertension and COVID-19?. Am J Hypertens. 2021;34(4):304–306.
- Sidarta-Oliveira D, Jara CP, Ferruzzi AJ, et al. SARS-CoV-2 receptor is co-expressed with elements of the Kinin-Kallikrein, renin-angiotensin and coagulation systems in alveolar cells. Sci Rep. 2020;10(1):19522.
- Gouda AS, Mégarbane B. Snake venom-derived bradykinin-potentiating peptides: a promising therapy for COVID-19? Drug Dev Res. 2021;82(1):38–48.
- Kaplan AP, Ghebrehiwet B. The plasma bradykinin-forming pathways and its interrelationships with complement. Mol Immunol. 2010;47(13):2161–2169.
- Marceau F, Bachelard H, Bouthillier J, et al. Bradykinin receptors: Agonists, antagonists, expression, signaling, and adaptation to sustained stimulation. Int Immunopharmacol. 2020;82(106305):106305.
- Kakoki M, Smithies O. The Kallikreinkinin system in health and in diseases of the kidney. Kidney Int. 2009;75(10):1019–1030.
- Zwaveling S, Gerth van Wijk R, Karim F. Pulmonary edema in COVID-19: explained by bradykinin? J Allergy Clin Immunol. 2020;146(6):1454–1455.
- Hoevenaar M, Goossens D, Roorda J. Angiotensin-converting enzyme 2, the complement system, the kallikrein-kinin system, type-2 diabetes, interleukin-6, and their interactions regarding the complex COVID-19 pathophysiological crossroads. J Renin Angiotensin Aldosterone Syst. 2020;21(4):1470320320979097.
- Lopatko Fagerström I, Ståhl A-L, Mossberg M, et al. Blockade of the kallikrein-kinin system reduces endothelial complement activation in vascular inflammation. EBioMedicine. 2019;47:319–328.
- Hofman Z, de Maat S, Hack CE, et al. Bradykinin: Inflammatory product of the coagulation system. Clin Rev Allergy Immunol. 2016;51(2):152–161.
- Qadri F, Bader M. Kinin B1 receptors as a therapeutic target for inflammation. Expert Opin Ther Targets. 2018;22(1):31–44.
- de Maat S, de Mast Q, Danser AHJ, et al. Impaired breakdown of bradykinin and its metabolites as a possible cause for pulmonary edema in COVID-19 infection. Semin Thromb Hemost. 2020;46(7):835–837.
- Oehmcke-Hecht S, Köhler J. Interaction of the human contact system with pathogens-an update. Front Immunol. 2018;9:312.
- Sodhi CP, Wohlford-Lenane C, Yamaguchi Y, et al. Attenuation of pulmonary ACE2 activity impairs inactivation of des-arg9 bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am J Physiol Lung Cell Mol Physiol. 2018;314(1):L17–L31.
- Wang G, Ye Y, Zhang X, et al. Bradykinin stimulates IL-6 production and cell invasion in colorectal cancer cells. Oncol Rep. 2014;32(4):1709–1714.
- Al-Shamlan F, El-Hashim AZ. Bradykinin sensitizes the cough reflex via a B2 receptor receptor receptor receptor dependent activation of TRPV1 and TRPA1 channels through metabolites of cyclooxygenase and 12-lipoxygenase. Respir Res. 2019;20(1):110.
- Ricciardolo FLM, Folkerts G, Folino A, et al. Bradykinin in asthma: modulation of airway inflammation and remodelling. Eur J Pharmacol. 2018;827:181–188.
- Karamyan VT. Between two storms, vasoactive peptides or bradykinin underlie severity of COVID-19? Physiol Rep. 2021;9(5):e14796.
- Ancion A, Tridetti J, Nguyen Trung ML, et al. A review of the role of bradykinin and nitric oxide in the cardioprotective action of angiotensin-converting enzyme inhibitors: Focus on perindopril. Cardiol Ther. 2019;8(2):179–191.
- Yang P, Kuc RE, Brame AL, et al. [Pyr1]apelin-13(1-12) is a biologically active ACE2 metabolite of the endogenous cardiovascular peptide [Pyr1]apelin-13. Front Neurosci. 2017;11:92.
- Shin K, Kenward C, Rainey JK. Apelinergic system structure and function. Compr Physiol. 2017;8(1):407–450.
- Chapman NA, Dupré DJ, Rainey JK. The apelin receptor: physiology, pathology, cell signalling, and ligand modulation of a peptide-activated class a GPCR. Biochem Cell Biol. 2014;92(6):431–440.
- Sato T, Suzuki T, Watanabe H, et al. Apelin is a positive regulator of ace2 in failing hearts. J Clin Invest. 2013;123(12):5203–5211.
- Wang W, Shen M, Fischer C, et al. Apelin protects against abdominal aortic aneurysm and the therapeutic role of neutral endopeptidase resistant apelin analogs. Proc Natl Acad Sci USA. 2019;116(26):13006–13015.
- Siddiquee K, Hampton J, McAnally D, et al. The apelin receptor inhibits the angiotensin II type 1 receptor via allosteric trans-inhibition. Br J Pharmacol. 2013;168(5):1104–1117.
- Masoud AG, Lin J, Azad AK, et al. Apelin directs endothelial cell differentiation and vascular repair following immune-mediated injury. J Clin Invest. 2020;130(1):94–107.
- Fan XF, Xue F, Zhang YQ, et al. The apelin-APJ axis is an endogenous counterinjury mechanism in experimental acute lung injury. Chest. 2015;147(4):969–978.
- Han S, Englander EW, Gomez GA, et al. Pancreatitis activates pancreatic apelin-APJ axis in mice. Am J Physiol Gastrointest Liver Physiol. 2013;305(2):G139–G150.
- Zhang H, Chen S, Zeng M, et al. Apelin-13 administration protects against LPS-induced acute lung injury by inhibiting NF-κB pathway and NLRP3 inflammasome activation. Cell Physiol Biochem. 2018;49(5):1918–1932.
- Yan J, Wang A, Cao J, et al. Apelin/APJ system: an emerging therapeutic target for respiratory diseases. Cell Mol Life Sci. 2020;77(15):2919–2930.
- Saeedi Saravi SS, Beer JH. Apelin-potential therapy for COVID-19? J Mol Cell Cardiol. 2020;145:84–87.
- Moriguchi T, Takai J. Histamine and histidine decarboxylase: immunomodulatory functions and regulatory mechanisms. Genes Cells. 2020;25(7):443–449.
- Tiligada E, Ennis M. Histamine pharmacology: from sir henry dale to the 21st century. Br J Pharmacol. 2020;177(3):469–489.
- Blasco MP, Chauhan A, Honarpisheh P, et al. Age-dependent involvement of gut mast cells and histamine in post-stroke inflammation. J Neuroinflammation. 2020;17:160. DOI:10.1186/s12974-020-01833-1
- Thangam EB, Jemima EA, Singh H, et al. The role of histamine and histamine receptors in mast cell-mediated allergy and inflammation: the hunt for new therapeutic targets. Front Immunol. 2018;9:1873.
- Branco ACCC, Yoshikawa FSY, Pietrobon AJ, et al. Role of histamine in modulating the immune response and inflammation. Mediators Inflamm. 2018;2018:9524075.
- Conti P, Caraffa A, Tetè G, et al. Mast cells activated by SARS-CoV-2 release histamine which increases IL-1 levels causing cytokine storm and inflammatory reaction in COVID-19. J Biol Regul Homeost Agents. 2020;34(5):1629–1632.
- Yamauchi K, Ogasawara M. The role of histamine in the pathophysiology of asthma and the clinical efficacy of antihistamines in asthma therapy. IJMS. 2019;20(7):1733.
- Mather JF, Seip RL, McKay RG. Impact of famotidine use on clinical outcomes of hospitalized patients with COVID-19. Am J Gastroenterol. 2020;115(10):1617–1623.
- Ge S, Wang X, Hou Y, et al. Repositioning of histamine H1 receptor antagonist: doxepin inhibits viropexis of SARS-CoV-2 spike Pseudovirus by blocking ACE2. Eur J Pharmacol. 2021;896:173897.
- Ennis M, Tiligada K. Histamine receptors and COVID-19. Inflamm Res. 2021;70(1):67–75.
- D’amico S, Tempora P, Lucarini V, et al. ERAP1 and ERAP2 enzymes: a protective shield for RAS against COVID-19? IJMS. 2021;22(4):1705.
- Saulle I, Vicentini C, Clerici M, et al. An overview on ERAP roles in infectious diseases. Cells. 2020;9(3):720.
- López De Castro JA. How ERAP1 and ERAP2 shape the peptidomes of disease-associated MHC-I proteins. Front Immunol. 2018;9:2463.
- Evnouchidou I, van Endert P. Peptide trimming by endoplasmic reticulum aminopeptidases: role of MHC class I binding and ERAP dimerization. Hum Immunol. 2019;80(5):290–295.
- Saulle I, Vanetti C, Goglia S, et al. A new ERAP2/Iso3 isoform expression is triggered by different microbial stimuli in human cells. could it play a role in the modulation of SARS-CoV-2 infection? Cells. 2020;9(9):1951.
- Stamatakis G, Samiotaki M, Mpakali A, et al. Generation of SARS-CoV-2 S1 spike glycoprotein putative antigenic epitopes in vitro by intracellular aminopeptidases. J Proteome Res. 2020;19(11):4398–4406.
- Reeves E, James E. The role of polymorphic ERAP1 in autoinflammatory disease. Biosci Rep. 2018;38(4):BSR20171503.
- Birra D, Benucci M, Landolfi L, et al. COVID 19: a clue from innate immunity. Immunol Res. 2020;68(3):161–168.
- Herrada AA, Escobedo N, Iruretagoyena M, et al. Innate immune cells’ contribution to systemic lupus erythematosus. Front Immunol. 2019;10:772.
- Romo MR, Pérez-Martínez D, Castillo Ferrer C. Innate immunity in vertebrates: an overview. Immunology. 2016;148(2):125–139.
- Guo Y-R, Cao Q-D, Hong Z-S, et al. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak – an update on the status. Mil Med Res. 2020;7(1):11.
- Angelopoulou A, Alexandris N, Konstantinou E, et al. Imiquimod – a toll like receptor 7 agonist – is an ideal option for management of COVID 19. Environ Res. 2020;188:109858.
- Brandão SCS, Ramos JdO, Dompieri LT, et al. Is toll-like receptor 4 involved in the severity of COVID-19 pathology in patients with cardiometabolic comorbidities? Cytokine Growth Factor Rev. 2021;58:102–110.
- Zheng M, Karki R, Williams EP, et al. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat Immunol. 2021;22(7):829–838.
- Carty M, Guy C, Bowie AG. Detection of viral infections by innate immunity. Biochem Pharmacol. 2021;183:114316.
- Menezes MCS, Veiga ADM, Martins de Lima T, et al. Lower peripheral blood toll-like receptor 3 expression is associated with an unfavorable outcome in severe COVID-19 patients. Sci Rep. 2021;11(1):152223.
- Choudhury A, Mukherjee S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J Med Virol. 2020;92(10):2105–2113.
- Shirato K, Kizaki T. SARS-CoV-2 spike protein S1 subunit induces pro-inflammatory responses via toll-like receptor 4 signaling in murine and human macrophages. Heliyon. 2021;7(2):e06187.
- Rebendenne A, Chaves Valadão AL, Tauziet M, et al. SARS-CoV-2 triggers an MDA-5-dependent interferon response which is unable to control replication in lung epithelial cells. J Virol. 2021;95(8):e02415–e02420.
- Yin X, Riva L, Pu Y, et al. MDA5 governs the innate immune response to SARS-CoV-2 in lung epithelial cells. Cell Rep. 2021;34(2):108628.
- Chow KT, Gale M, Loo YM. RIG-I and other RNA sensors in antiviral immunity. Annu Rev Immunol. 2018;36:667–694.
- Yamada T, Sato S, Sotoyama Y, et al. RIG-I triggers a signaling-abortive anti-SARS-CoV-2 defense in human lung cells. Nat Immunol. 2021;22(7):820–828.
- McCarty MF, Assanga SBI, Luján LL, et al. Nutraceutical strategies for suppressing nlrp3 inflammasome activation: Pertinence to the management of covid-19 and beyond. Nutrients. 2020;13(1):47.
- Pan P, Shen M, Yu Z, et al. SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat Commun. 2021;12:4664.
- Paniri A, Akhavan-Niaki H. Emerging role of IL-6 and NLRP3 inflammasome as potential therapeutic targets to combat COVID-19: Role of lncRNAs in cytokine storm modulation. Life Sci. 2020;257:118114.
- Moretti J, Blander JM. Increasing complexity of NLRP3 inflammasome regulation. J Leukoc Biol. 2021;109(3):561–571.
- Kolbrink B, Riebeling T, Kunzendorf U, et al. Plasma membrane pores drive inflammatory cell death. Front Cell Dev Biol. 2020;8:817.
- Ratajczak MZ, Bujko K, Ciechanowicz A, et al. SARS-CoV-2 entry receptor ACE2 is expressed on very small CD45- precursors of hematopoietic and endothelial cells and in response to virus spike protein activates the Nlrp3 inflammasome. Stem Cell Rev Rep. 2021;17(1):266–277.
- Abdullah FS, Aman K, ur Rahman A. Innate immune-mediated antiviral response to SARS-CoV-2 and convalescent sera a potential prophylactic and therapeutic agent to tackle COVID-19. Antib Ther. 2020;3(3):213–220.
- Hasselbalch HC, Skov V, Kjaer L, et al. COVID-19 as a mediator of interferon deficiency and hyperinflammation: rationale for the use of JAK1/2 inhibitors in combination with interferon. Cytokine Growth Factor Rev. 2021;60:28–45.
- Mesev EV, LeDesma RA, Ploss A. Decoding type I and III interferon signalling during viral infection. Nat Microbiol. 2019;4(6):914–924.
- Goker Bagca B, Biray Avci C. The potential of JAK/STAT pathway inhibition by ruxolitinib in the treatment of COVID-19. Cytokine Growth Factor Rev. 2020;54:51–61.
- Seif F, Aazami H, Khoshmirsafa M, et al. JAK inhibition as a new treatment strategy for patients with COVID-19. Int Arch Allergy Immunol. 2020;181(6):467–475.
- Choudhary S, Sharma K, Silakari O. The interplay between inflammatory pathways and COVID-19: a critical review on pathogenesis and therapeutic options. Microb Pathog. 2021;150:104673.
- Karki R, Sharma BR, Tuladhar S, et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell. 2021;184(1):149–168.
- Laing AG, Lorenc A, Molino del Barrio I D, et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat Med. 2020;26(10):1623–1635.
- Galani IE, Rovina N, Lampropoulou V, et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat Immunol. 2021;22(1):32–40.
- Chen X, Zhao B, Qu Y, et al. Detectable serum severe acute respiratory syndrome coronavirus 2 viral load (RNAemia) is closely correlated with drastically elevated interleukin 6 level in critically ill patients with coronavirus disease 2019. Clin Infect Dis. 2020;71(8):1937–1942.
- Cavalli G, Larcher A, Tomelleri A, et al. Interleukin-1 and interleukin-6 inhibition compared with standard management in patients with COVID-19 and hyperinflammation: a cohort study. Lancet Rheumatol. 2021;3(4):e253–e261.
- Gubernatorova EO, Gorshkova EA, Polinova AI, et al. IL-6: relevance for immunopathology of SARS-CoV-2. Cytokine Growth Factor Rev. 2020;53:13–24.
- Gómez-rial J, Rivero-calle I, Salas A, et al. Role of monocytes/macrophages in covid-19 pathogenesis: implications for therapy. Infect Drug Resist. 2020;13:2485–2493.
- Gando S, Wada T. Thromboplasminflammation in COVID-19 coagulopathy: three viewpoints for diagnostic and therapeutic strategies. Front Immunol. 2021;12:649122.
- Bautista-Vargas M, Bonilla-Abadía F, Cañas CA. Potential role for tissue factor in the pathogenesis of hypercoagulability associated with in COVID-19. J Thromb Thrombolysis. 2020;50(3):479–483.
- Quartuccio L, Fabris M, Sonaglia A, et al. Interleukin 6, soluble interleukin 2 receptor alpha (CD25), monocyte colony-stimulating factor, and hepatocyte growth factor linked with systemic hyperinflammation, innate immunity hyperactivation, and organ damage in COVID-19 pneumonia. Cytokine. 2021;140:155438.
- Di Spigna G, Spalletti Cernia D, Vargas M, et al. Drastically elevated levels of interleukin-6 and its soluble receptor complex in COVID-19 patients with acute respiratory distress. Clin Med Invest. 2020;5(3):1–4.
- Chen LYC, Biggs CM, Jamal S, et al. Soluble interleukin-6 receptor in the COVID-19 cytokine storm syndrome. Cell Rep Med. 2021;2(5):100269.
- McGonagle D, Sharif K, O'Regan A, et al. The role of cytokines including interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun Rev. 2020;19(6):102537.
- Rabaan AA, Al-Ahmed SH, Muhammad J, et al. Role of inflammatory cytokines in covid-19 patients: a review on molecular mechanisms, immune functions, immunopathology and immunomodulatory drugs to counter cytokine storm. Vaccines. 2021;9(5):436.
- Zheng J, Wang Y, Li K, et al. Severe acute respiratory syndrome coronavirus 2-Induced immune activation and death of monocyte-derived human macrophages and dendritic cells. J Infect Dis. 2021;223(5):785–795.
- Toor SM, Saleh R, Sasidharan Nair V, et al. T-cell responses and therapies against SARS-CoV-2 infection. Immunology. 2021;162(1):30–43.
- Palacios Y, Ruiz A, Ramón-Luing LA, et al. Severe COVID-19 patients show an increase in soluble TNFR1 and ADAM17, with a relationship to mortality. IJMS. 2021;22(16):8423.
- Ahmad S, Hatmal MM, Lambuk L, et al. The role of TNFR2+ tregs in COVID-19: an overview and a potential therapeutic strategy. Life Sci. 2021;286:120063.
- Janiuk K, Jabłońska E, Garley M. Significance of NETs formation in COVID-19. Cells. 2021;10(1):151.
- Liberale L, Holy EW, Akhmedov A, et al. Interleukin-1β mediates arterial thrombus formation via NET-associated tissue factor. JCM. 2019;8(12):2072.
- Geng J, Wang F, Huang Z, et al. Perspectives on anti-IL-1 inhibitors as potential therapeutic interventions for severe COVID-19. Cytokine. 2021;143:155544.
- Lei X, Dong X, Ma R, et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun. 2020;11(1):3810.
- Banerjee A, El-Sayes N, Budylowski P, et al. Experimental and natural evidence of SARS-CoV-2-infection-induced activation of type I interferon responses. iScience. 2021;24(5):102477.
- Lucas C, Wong P, Klein J, Yale IMPACT Team, et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature. 2020;584(7821):463–469.
- Broggi A, Ghosh S, Sposito B, et al. Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science. 2020;369(6504):706–712.
- Busnadiego I, Fernbach S, Pohl MO, et al. Antiviral activity of type i, ii, and iii interferons counterbalances ace2 inducibility and restricts SARS-CoV-2. MBio. 2020;11(5):e01928–20.
- Ahn D, Prince A. Participation of the IL-10RB related cytokines, IL-22 and IFN-λ in defense of the airway mucosal barrier. Front Cell Infect Microbiol. 2020;10:300.
- Smieszek SP, Polymeropoulos VM, Xiao C, et al. Loss-of-function mutations in IFNAR2 in COVID-19 severe infection susceptibility. J Glob Antimicrob Resist. 2021;26:239–240.
- Velavan TP, Pallerla SR, Rüter J, et al. Host genetic factors determining COVID-19 susceptibility and severity. EBioMedicine. 2021;72:103629.
- Kim YM, Shin EC. Type I and III interferon responses in SARS-CoV-2 infection. Exp Mol Med. 2021;53(5):750–760.
- Zhou Z, Ren L, Zhang L, et al. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe. 2020;27(6):883–890.
- Xiong Y, Liu Y, Cao L, et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg Microbes Infect. 2020;9(1):761–770.
- Blanco-Melo D, Nilsson-Payant BE, Liu WC, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181(5):1036–1045.
- Yang Y, Shen C, Li J, et al. Plasma IP-10 and MCP-3 levels are highly associated with disease severity and predict the progression of COVID-19. J Allergy Clin Immunol. 2020;146(1):119–127.
- Yang L, Xie X, Tu Z, et al. The signal pathways and treatment of cytokine storm in COVID-19. Signal Transduct Target Ther. 2021;6(1):255.
- Kim JS, Lee JY, Yang JW, et al. Immunopathogenesis and treatment of cytokine storm in COVID-19. Theranostics. 2021;11(1):316–329.
- Tan LY, Komarasamy TV, Balasubramaniam R, et al. Immune response and COVID-19: a double edged sword. Front Immunol. 2021;12:742941.
- Hadjadj J, Yatim N, Barnabei L, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369(6504):718–724.
- Prokunina-Olsson L, Alphonse N, Dickenson RE, et al. COVID-19 and emerging viral infections: the case for interferon lambda. J Exp Med. 2020;217(5):e20200653.
- Major J, Crotta S, Llorian M, et al. Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science. 2020;369(6504):712–717.
- Vanderheiden A, Ralfs P, Chirkova T, et al. Type I and type III interferons restrict SARS-CoV-2 infection of human airway epithelial cultures. J Virol. 2020;94(19):e00985.
- Yuen C, Lam J, Wong W, et al. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg Microbes Infect. 2020;9(1):1418–1428.
- Miorin L, Kehrer T, Sanchez-Aparicio MT, et al. SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc Natl Acad Sci USA. 2020;117(45):28344–28354.
- Rui Y, Su J, Shen S, et al. Unique and complementary suppression of cGAS-STING and RNA sensing- triggered innate immune responses by SARS-CoV-2 proteins. Signal Transduct Target Ther. 2021;6(1):123.
- Han L, Zhuang MW, Deng J, et al. SARS-CoV-2 ORF9b antagonizes type I and III interferons by targeting multiple components of the RIG-I/MDA-5–MAVS, TLR3–TRIF, and cGAS–STING signaling pathways. J Med Virol. 2021;93(9):5376–5389.
- England JT, Abdulla A, Biggs CM, et al. Weathering the COVID-19 storm: lessons from hematologic cytokine syndromes. Blood Rev. 2021;45:100707.
- Meidaninikjeh S, Sabouni N, Marzouni HZ, et al. Monocytes and macrophages in COVID-19: friends and foes. Life Sci. 2021;269(119010):119010.
- Merad M, Martin JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;20(6):355–362.
- Wauters E, Van Mol P, Garg AD, CONTAGIOUS orators, et al. Discriminating mild from critical COVID-19 by innate and adaptive immune single-cell profiling of bronchoalveolar lavages. Cell Res. 2021;31(3):272–290.
- Gustine JN, Jones D. Immunopathology of hyperinflammation in COVID-19. Am J Pathol. 2021;191(1):4–17.
- Moore JB, June CH. Cytokine release syndrome in severe COVID-19. Science. 2020;368(6490):473–474.
- Otsuka R, Seino KI. Macrophage activation syndrome and COVID-19. Inflamm Regen. 2020;40(19):19.
- Mehta P, McAuley DF, Brown M, HLH Across Speciality Collaboration, UK, et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395(10229):1033–1034.
- Eslamifar Z, Behzadifard M, Soleimani M, et al. Coagulation abnormalities in SARS-CoV-2 infection: overexpression tissue factor. Thromb J. 2020;18(1):38.
- Wang C, Xie J, Zhao L, et al. Alveolar macrophage dysfunction and cytokine storm in the pathogenesis of two severe COVID-19 patients. EBioMedicine. 2020;57:102833.
- Jafarzadeh A, Chauhan P, Saha B, et al. Contribution of monocytes and macrophages to the local tissue inflammation and cytokine storm in COVID-19: lessons from SARS and MERS, and potential therapeutic interventions. Life Sci. 2020;257:118102.
- Yang D, Chu H, Hou Y, et al. Attenuated interferon and proinflammatory response in SARS-CoV-2-infected human dendritic cells is associated with viral antagonism of STAT1 phosphorylation. J Infect Dis. 2020;222(5):734–745.
- Gracia-Hernandez M, Sotomayor EM, Villagra A. Targeting macrophages as a therapeutic option in coronavirus disease. Front Pharmacol. 2019;11:577571.
- Desai N, Neyaz A, Szabolcs A, et al. Temporal and spatial heterogeneity of host response to SARS-CoV-2 pulmonary infection. Nat Commun. 2020;11:6319. DOI:10.1038/s41467-020-20139-7
- Matic S, Popovic S, Djurdjevic P, et al. SARS-CoV-2 infection induces mixed M1/M2 phenotype in circulating monocytes and alterations in both dendritic cell and monocyte subsets. PLOS One. 2020;15(12):e0241097.
- Pence BD. Severe COVID-19 and aging: are monocytes the key? Geroscience. 2020;42(4):1051–1061.
- Borges L, Pithon-Curi TC, Curi R, et al. COVID-19 and neutrophils: the relationship between hyperinflammation and neutrophil extracellular traps. Mediators Inflamm. 2020;2020:1–7.
- Liew PX, Kubes P. The neutrophil's role during health and disease. Physiol Rev. 2019;99(2):1223–1248.
- Dhama K, Patel SK, Pathak M, et al. An update on SARS-CoV-2/COVID-19 with particular reference to its clinical pathology, pathogenesis, immunopathology and mitigation strategies. Travel Med Infect Dis. 2020;37:101755.
- Wang J, Li Q, Yin Y, et al. Excessive neutrophils and neutrophil extracellular traps in COVID-19. Front Immunol. 2020;11:2063.
- Hazeldine J, Lord JM. Neutrophils and COVID-19: active participants and rational therapeutic targets. Front Immunol. 2021;12:680134.
- Cavalcante-Silva LHA, Carvalho DCM, Lima É de A, et al. Neutrophils and COVID-19: the road so far. Int Immunopharmacol. 2021;90:107233.
- Veras F, Pontelli M, Silva C, et al. SARS-CoV-2 triggered neutrophil extracellular traps (NETs) mediate COVID-19 pathology. J Exp Med. 2020;217(12):e20201129.
- Tomar B, Anders H-J, Desai J, et al. Neutrophils and neutrophil extracellular traps drive necroinflammation in COVID-19. Cells. 2020;9(6):1383.
- Thierry AR, Roch B. Neutrophil extracellular traps and by-products play a key role in COVID-19: pathogenesis, risk factors, and therapy. JCM. 2020;9(9):2942.
- Middleton EA, He XY, Denorme F, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. 2020;136(10):1169–1179.
- Ng H, Havervall S, Rosell A, et al. Circulating markers of neutrophil extracellular traps are of prognostic value in patients with COVID-19. Arterioscler Thromb Vasc Biol. 2021;41(2):988–994.
- Ge Y, Huang M, Yao YM. Biology of interleukin-17 and its pathophysiological significance in sepsis. Front Immunol. 2020;11:1558.
- Zhang Y, Chandra V, Sanchez ER, et al. Interleukin-17-induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J Exp Med. 2020;217(12):e20190354.
- Maxwell AJ, Ding J, You Y, et al. Identification of key signaling pathways induced by SARS-CoV2 that underlie thrombosis and vascular injury in COVID-19 patients. J Leukoc Biol. 2021;109(1):35–47.
- Joncour A, Le Biard L, Vautier M, et al. Neutrophil – platelet and monocyte – platelet aggregates in COVID-19 patients. Thromb Haemost. 2020;120(12):1733–1735.
- Lindsley AW, Schwartz JT, Rothenberg ME. Eosinophil responses during COVID-19 infections and coronavirus vaccination. J Allergy Clin Immunol. 2020;146(1):1–7.
- Weller PF, Spencer LA. Functions of tissue-resident eosinophils. Nat Rev Immunol. 2017;17(12):746–760.
- Williams TL, Rada B, Tandon E, et al. Nets and EETs, a whole web of. Mess. Microorganisms. 2020;8(12):1925.
- Koenderman L, Siemers MJ, van Aalst C, et al. The systemic immune response in COVID-19 is associated with a shift to formyl-peptide unresponsive eosinophils. Cells. 2021;10(5):1109.
- P Le B, Vuillaume LA, Alamé K, et al. Do blood eosinophils predict in-hospital mortality or severity of disease in SARS-CoV-2 infection? A retrospective multicenter study. Microorganisms. 2021;9(2):334.
- Valverde-Monge M, Cañas JA, Barroso B, et al. Eosinophils and chronic respiratory diseases in hospitalized COVID-19 patients. Front Immunol. 2021;12:668074.
- Rosenberg HF, Foster PS. Eosinophils and COVID-19: diagnosis, prognosis, and vaccination strategies. Semin Immunopathol. 2021;43(3):383–392.
- Rodriguez L, Pekkarinen PT, Lakshmikanth T, et al. Systems-Level immunomonitoring from acute to recovery phase of severe COVID-19. Cell Rep Med. 2020;1(5):100078.
- Kanagaratham C, El Ansari YS, Lewis OL, et al. IgE and IgG antibodies as regulators of mast cell and basophil functions in food allergy. Front Immunol. 2020;11:603050.
- Vitte J, Diallo AB, Boumaza A, et al. A granulocytic signature identifies COVID-19 and its severity. J Infect Dis. 2020;222(12):1985–1996.
- Renner K, Schwittay T, Chaabane S, et al. Severe T cell hyporeactivity in ventilated COVID-19 patients correlates with prolonged virus persistence and poor outcomes. Nat Commun. 2021;12:3006. DOI:10.1038/s41467-021-23334-2
- Sun Y, Zhou J, Ye K. White blood cells and severe covid-19: a mendelian randomization study. JPM. 2021;11(3):195.
- Market M, Angka L, Martel AB, et al. Flattening the COVID-19 curve with natural killer cell based immunotherapies. Front Immunol. 2020;11:1512.
- Björkström NK, Strunz B, Ljunggren H-G. Natural killer cells in antiviral immunity. Nat Rev Immunol. 2022;22(2):112–123.
- Diaz-Salazar C, Sun JC. Natural killer cell responses to emerging viruses of zoonotic origin. Curr Opin Virol. 2020;44:97–111.
- Bao C, Tao X, Cui W, et al. Natural killer cells associated with SARS-CoV-2 viral RNA shedding, antibody response and mortality in COVID-19 patients. Exp Hematol Oncol. 2021;10(1):5.
- Zuo W, Zhao X. Natural killer cells play an important role in virus infection control: antiviral mechanism, subset expansion and clinical application. Clin Immunol. 2021;227:108727.
- Giamarellos-Bourboulis EJ, Netea MG, Rovina N, et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe. 2020;27(6):992–1000.
- Yang B, Fan J, Huang J, et al. Clinical and molecular characteristics of COVID-19 patients with persistent SARS-CoV-2 infection. Nat Commun. 2021;12:3501. DOI:10.1038/s41467-021-23621-y
- Mazzoni A, Salvati L, Maggi L, et al. Impaired immune cell cytotoxicity in severe COVID-19 is IL-6 dependent. J Clin Invest. 2020;130(9):4694–4703.
- Masselli E, Vaccarezza M, Carubbi C, et al. NK cells: a double edge sword against SARS-CoV-2. Adv Biol Regul. 2020;77:100737.
- Bozzano F, Dentone C, Perrone C, GECOVID study group, et al. Extensive activation, tissue trafficking, turnover and functional impairment of NK cells in COVID-19 patients at disease onset associates with subsequent disease severity. PLoS Pathog. 2021;17(4):e1009448.
- Antonioli L, Fornai M, Pellegrini C, et al. NKG2A and COVID-19: another brick in the wall. Cell Mol Immunol. 2020;17(6):672–674.
- Demaria O, Carvelli J, Batista L, et al. Identification of druggable inhibitory immune checkpoints on natural killer cells in COVID-19. Cell Mol Immunol. 2020;17(9):995–997.
- Bortolotti D, Gentili V, Rizzo S, et al. SARS-CoV-2 spike 1 protein controls natural killer cell activation via the HLA-E/NKG2A pathway. Cell. 2020;9(9):1975.
- Kempuraj D, Selvakumar GP, Ahmed ME, et al. COVID-19, mast cells, cytokine storm, psychological stress, and neuroinflammation. Neuroscientist. 2020;26(5–6):402–414.
- Abdin SM, Elgendy SM, Alyammahi SK, et al. Tackling the cytokine storm in COVID-19, challenges and hopes. Life Sci. 2020;257:118054.
- Gigante A, Aquili A, Farinelli L, et al. Sodium chromo-glycate and palmitoylethanolamide: a possible strategy to treat mast cell-induced lung inflammation in COVID-19. Med Hypotheses. 2020;143:109856.
- Theoharides TC. Potential association of mast cells with coronavirus disease 2019. Ann Allergy Asthma Immunol. 2021;126(3):217–218.
- Gebremeskel S, Schanin J, Coyle KM, et al. Mast cell and eosinophil activation are associated with COVID-19 and TLR-mediated viral inflammation: Implications for an anti-Siglec-8 antibody. Front Immunol. 2021;12:650331.
- Junior JdS, Miggiolaro AdS, Nagashima S, et al. Mast cells in alveolar septa of COVID-19 patients: a pathogenic pathway that may link interstitial edema to immunothrombosis. Front Immunol. 2020;11:574862.
- Han J, Sun J, Zhang G, et al. Dcs-based therapies: potential strategies in severe sars-cov-2 infection. Int J Med Sci. 2021;18(2):406–418.
- Alamri A, Fisk D, Upreti D, et al. A missing link: engagements of dendritic cells in the pathogenesis of SARS-CoV-2 infections. IJMS. 2021;22(3):1118.
- Campana P, Parisi V, Leosco D, et al. A. Dendritic cells and SARS-CoV-2 infection: still an unclarified connection. Cells. 2020;9(9):2046.
- Döbel T, Schäkel K. The role of human 6-sulfo LacNAc dendritic cells (slanDCs) in autoimmunity and tumor diseases. J Der Dtsch Dermatologischen Gesellschaft. 2014;12(10):874–879.
- Wilk AJ, Rustagi A, Zhao NQ, et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med. 2020;26(7):1070–1076.
- Peruzzi B, Bencini S, Capone M, et al. Quantitative and qualitative alterations of circulating myeloid cells and plasmacytoid DC in SARS-CoV-2 infection. Immunology. 2020;161(4):345–353.
- Zingaropoli MA, Nijhawan P, Carraro A, et al. Increased sCD163 and sCD14 plasmatic levels and depletion of peripheral blood Pro-Inflammatory monocytes, myeloid and plasmacytoid dendritic cells in patients with severe COVID-19 pneumonia. Front Immunol. 2021;12:627548.
- Zhou R, To KKW, Wong YC, et al. Acute SARS-CoV-2 infection impairs dendritic cell and T cell responses. Immunity. 2020;53(4):864–877.
- Shi W, Liu X, Cao Q, et al. High-dimensional single-cell analysis reveals the immune characteristics of COVID-19. Am J Physiol Lung Cell Mol Physiol. 2021;320(1):L84–L98.
- Buttenschön J, Mattner J. The interplay between dendritic cells and CD8 T lymphocytes is a crucial component of SARS-CoV-2 immunity. Cell Mol Immunol. 2021;18(2):247–249.
- Kumar V. How could we forget immunometabolism in SARS-CoV2 infection or COVID-19? Int Rev Immunol. 2021;40(1–2):72–107.
- Onodi F, Bonnet-Madin L, Meertens L, et al. SARS-CoV-2 induces human plasmacytoid predendritic cell diversification via UNC93B and IRAK4. J Exp Med. 2021;218(4):e20201387.
- Xiang Q, Feng Z, Diao B, et al. SARS-CoV-2 induces lymphocytopenia by promoting inflammation and decimates secondary lymphoid organs. Front Immunol. 2021;12:661052.
- Niles MA, Gogesch P, Kronhart S, et al. Macrophages and dendritic cells are not the major source of Pro-Inflammatory cytokines upon SARS-CoV-2 infection. Front Immunol. 2021;12:647824.
- Yousif AS, Ronsard L, Shah P, et al. The persistence of interleukin-6 is regulated by a blood buffer system derived from dendritic cells. Immunity. 2021;54(2):235–246. e5.
- McElvaney OJ, Curley GF, Rose-John S, et al. Interleukin-6: obstacles to targeting a complex cytokine in critical illness. Lancet Respir Med. 2021;9(6):643–654.
- Saadeldin MK, Abdel-Aziz AK, Abdellatif A. Dendritic cell vaccine immunotherapy; the beginning of the end of cancer and COVID-19. A hypothesis. Med Hypotheses. 2021;146:110365.
- Golchin A. Cell-Based therapy for severe COVID-19 patients. Stem Cell Rev Rep. 2021;17(1):56–62.
- Marchetti M. COVID-19-driven endothelial damage: complement, HIF-1, and ABL2 are potential pathways of damage and targets for cure. Ann Hematol. 2020;99(8):1701–1707.
- Java A, Apicelli AJ, Kathryn Liszewski M, et al. The complement system in COVID-19: friend and foe? JCI Insight. 2020;5(15):e140711.
- Ng N, Powell CA. Targeting the complement cascade in the pathophysiology of COVID-19 disease. JCM. 2021;10(10):2188.
- Stenmark KR, Frid MG, Gerasimovskaya E, et al. Mechanisms of SARS-CoV-2-induced lung vascular disease: potential role of complement. Pulm Circ. 2021;11(2):1–14.
- Yan B, Freiwald T, Chauss D, et al. SARS-CoV-2 drives JAK1/2-dependent local complement hyperactivation. Sci Immunol. 2021;6(58):eabg0833.
- Magro C, Mulvey JJ, Berlin D, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. 2020;220:1–13.
- Ma L, Sahu SK, Cano M, et al. Increased complement activation is a distinctive feature of severe SARS-CoV-2 infection. Sci Immunol. 2021;6(59):eabh2259.
- Satyam A, Tsokos MG, Brook OR, et al. Activation of classical and alternative complement pathways in the pathogenesis of lung injury in COVID-19. Clin Immunol. 2021;226:108716.
- Messner CB, Demichev V, Wendisch D, et al. Ultra-high-throughput clinical proteomics reveals classifiers of COVID-19 infection. Cell Syst. 2020;11(1):11–24.
- Yu J, Yuan X, Chen H, et al. Direct activation of the alternative complement pathway by SARS-CoV-2 spike proteins is blocked by factor D inhibition. Blood. 2020;136(18):2080–2089.
- Flude BM, Nannetti G, Mitchell P, et al. Targeting the complement serine protease masp-2 as a therapeutic strategy for coronavirus infections. Viruses. 2021;13(2):312.
- Bosmann M. Complement control for COVID-19. Sci Immunol. 2021;6(59):eabj1014.
- Sinkovits G, Mező B, Réti M, et al. Complement overactivation and consumption predicts in-hospital mortality in SARS-CoV-2 infection. Front Immunol. 2021;12:663187.
- de Nooijer AH, Grondman I, Janssen NAF, RCI-COVID-19 study group, et al. Complement activation in the disease course of coronavirus disease 2019 and its effects on clinical outcomes. J Infect Dis. 2021;223(2):214–224.
- Carvelli J, Demaria O, Vély F, Explore COVID-19 Marseille Immunopole group, et al. Association of COVID-19 inflammation with activation of the C5a-C5aR1 axis. Nature. 2020;588(7836):146–150.
- Cugno M, Meroni PL, Gualtierotti R, et al. Complement activation and endothelial perturbation parallel COVID-19 severity and activity. J Autoimmun. 2021;116:102560.
- Cheng W, Hornung R, Xu K, et al. Complement C3 identified as a unique risk factor for disease severity among young COVID-19 patients in Wuhan. China. Sci Rep. 2021;11:7857.
- Vlaar APJ, de Bruin S, Brouwer MC, et al. Efficacy matters: broadening complement inhibition in COVID-19 – authors' reply. Lancet Rheumatol. 2021;3(2):e95–e96.
- Vitiello A, La Porta R, D'Aiuto V, et al. F. Pharmacological approach for the reduction of inflammatory and prothrombotic hyperactive state in COVID-19 positive patients by acting on complement cascade. Hum Immunol. 2021;82(4):264–269.
- Skendros P, Mitsios A, Chrysanthopoulou A, et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Invest. 2020;130(11):6151–6157.
- Napoli C, Benincasa G, Criscuolo C, et al. Immune reactivity during COVID-19: implications for treatment. Immunol Lett. 2021;231:28–34.
- Kalfaoglu B, Almeida-Santos J, Tye CA, et al. T-cell dysregulation in COVID-19. Biochem Biophys Res Commun. 2021;538:204–210.
- Combadière B. Immunité adaptative contre le virus SARS-CoV-2. Med Sci. 2020;36(10):908–913.
- Hanna SJ, Codd AS, Gea-Mallorqui E, Oxford-Cardiff COVID-19 Literature Consortium, et al. T cell phenotypes in COVID-19 – a living review. Oxf Open Immunol. 2021;2(1):iqaa007.
- Mohamed Khosroshahi L, Rokni M, Mokhtari T, et al. Immunology, immunopathogenesis and immunotherapeutics of COVID-19; an overview. Int Immunopharmacol. 2021;93:107364.
- Sun H, Sun C, Xiao W, et al. Tissue-resident lymphocytes: from adaptive to innate immunity. Cell Mol Immunol. 2019;16(3):205–215.
- Jarjour NN, Masopust D, Jameson SC. T cell memory: understanding COVID-19. Immunity. 2021;54(1):14–18.
- Ni L, Ye F, Cheng ML, et al. Detection of SARS-CoV-2-Specific humoral and cellular immunity in COVID-19 convalescent individuals. Immunity. 2020;52(6):971–977.
- Tan AT, Linster M, Tan CW, et al. Early induction of functional SARS-CoV-2-specific T cells associates with rapid viral clearance and mild disease in COVID-19 patients. Cell Rep. 2021;34(6):108728.
- Bobcakova A, Petriskova J, Vysehradsky R, et al. Immune profile in patients with COVID-19: lymphocytes exhaustion markers in relationship to clinical outcome. Front Cell Infect Microbiol. 2021;11:646688.
- Schulte-Schrepping J, Reusch N, Paclik D, Deutsche COVID-19 OMICS Initiative (DeCOI), et al. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell. 2020;182(6):1419–1440.
- Yaqinuddin A, Kashir J. Novel therapeutic targets for SARS-CoV-2-induced acute lung injury: targeting a potential IL-1β/neutrophil extracellular traps feedback loop. Med Hypotheses. 2020;143:109906.
- Andonegui-Elguera S, Taniguchi-Ponciano K, Gonzalez-Bonilla CR, et al. Molecular alterations prompted by SARS-CoV-2 infection: Induction of hyaluronan, glycosaminoglycan and mucopolysaccharide metabolism. Arch Med Res. 2020;51(7):645–653.
- Yan B, Yang J, Xie Y, et al. Relationship between blood eosinophil levels and COVID-19 mortality. World Allergy Organ J. 2021;14(3):100521.
- Lv J, Wang Z, Qu Y, et al. Distinct uptake, amplification, and release of SARS-CoV-2 by M1 and M2 alveolar macrophages. Cell Discov. 2021;7(1):24.
- Mathew D, Giles JR, Baxter AE, The UPenn COVID Processing Unit, et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369(6508):eabc8511.
- Schultheiß C, Paschold L, Simnica D, et al. Next-generation sequencing of T and B cell receptor repertoires from COVID-19 patients showed signatures associated with severity of disease. Immunity. 2020;53(2):442–455.
- Grau-Expósito J, Sánchez-Gaona N, Massana N, et al. Peripheral and lung resident memory T cell responses against SARS-CoV-2. Nat Commun. 2021;12:3010. DOI:10.1038/s41467-021-23333-3
- Liao M, Liu Y, Yuan J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med. 2020;26(6):842–844.
- Rydyznski Moderbacher C, Ramirez SI, Dan JM, et al. Antigen-specific adaptive immunity to SARS-CoV-2 in acute COVID-19 and associations with age and disease severity. Cell. 2020;183(4):996–1012.
- Chen Z, Ruan P, Wang L, et al. T and B cell epitope analysis of SARS-CoV-2 S protein based on immunoinformatics and experimental research. J Cell Mol Med. 2021;25(2):1274–1289.
- Thieme CJ, Anft M, Paniskaki K, et al. Robust T cell response toward spike, membrane, and nucleocapsid SARS-CoV-2 proteins is not associated with recovery in critical COVID-19 patients. Cell Reports Med. 2020;1(6):100092.
- Gangaev A, Ketelaars SLC, Isaeva OI, et al. Identification and characterization of a SARS-CoV-2 specific CD8+ T cell response with immunodominant features. Nat Commun. 2021;12:2593. DOI:10.1038/s41467-021-22811-y
- Wang Z, Yang X, Zhong J, et al. Exposure to SARS-CoV-2 generates T-cell memory in the absence of a detectable viral infection. Nat Commun. 2021;12(1):6–13.
- Weiskopf D, Schmitz KS, Raadsen MP, et al. Phenotype and kinetics of SARS-CoV-2-specific T cells in COVID-19 patients with acute respiratory distress syndrome. Sci Immunol. 2020;5(48):eabd2071.
- Schulien I, Kemming J, Oberhardt V, et al. Characterization of pre-existing and induced SARS-CoV-2-specific CD8+ T cells. Nat Med. 2021;27(1):78–85.
- Bouadma L, Wiedemann A, Patrier J, et al. Immune alterations in a patient with SARS-CoV-2-related acute respiratory distress syndrome. J Clin Immunol. 2020;40(8):1082–1092.
- Gadotti AC, de Castro Deus M, Telles JP, et al. IFN-γ is an independent risk factor associated with mortality in patients with moderate and severe COVID-19 infection. Virus Res. 2020;289(198171):198171.
- Oja AE, Saris A, Ghandour CA, et al. Divergent SARS-CoV-2-specific T- and B-cell responses in severe but not mild COVID-19 patients. Eur J Immunol. 2020;50(12):1998–2012.
- Neidleman J, Luo X, Frouard J, et al. SARS-CoV-2-Specific T cells exhibit phenotypic features of helper function, lack of terminal differentiation, and high proliferation potential. Cell Rep Med. 2020;1(6):100081.
- Baumjohann D, Fazilleau N. Antigen-dependent multistep differentiation of T follicular helper cells and its role in SARS-CoV-2 infection and vaccination. Eur J Immunol. 2021;51(6):1325–1333.
- Riou C, Schäfer G, Bruyn E, et al. Rapid, simplified whole blood-based multiparameter assay to quantify and phenotype SARS-CoV-2 specific T cells. Eur Respir J. 2021;58(1):2100285.
- Jagannathan P, Wang TT. Immunity after SARS-CoV-2 infections. Nat Immunol. 2021;22(5):539–540.
- Dan JM, Mateus J, Kato Y, et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science (80-). 2021;371(6529):eabf4063.
- Sherina N, Piralla A, Du L, et al. Persistence of SARS-CoV-2-specific B and T cell responses in convalescent COVID-19 patients 6–8 months after the infection. Med. 2021;2(3):281–295.
- Rodda LB, Netland J, Shehata L, et al. Functional SARS-CoV-2-specific immune memory persists after mild COVID-19. Cell. 2021;184(1):169–183.
- Wang F, Hou H, Luo Y, et al. The laboratory tests and host immunity of COVID-19 patients with different severity of illness. JCI Insight. 2020;5(10):e137799.
- Gao M, Liu Y, Guo M, et al. Regulatory CD4+ and CD8+ T cells are negatively correlated with CD4+/CD8+ T cell ratios in patients acutely infected with SARS-CoV-2. J Leukoc Biol. 2021;109(1):91–97.
- Röltgen K, Boyd SD. Antibody and B cell responses to SARS-CoV-2 infection and vaccination. Cell Host Microbe. 2021;29(7):1063–1075.
- Detsika MG, Ampelakiotou K, Grigoriou E, et al. A novel ratio of CD8+:B-cells as a prognostic marker of coronavirus disease 2019 patient progression and outcome. Virology. 2021;556:79–86.
- Ihlow J, Michaelis E, Greuel S, et al. B cell depletion and signs of sepsis-acquired immunodeficiency in bone marrow and spleen of COVID-19 deceased. Int J Infect Dis. 2021;103:628–635.
- Liu Y, Tan W, Chen H, et al. Dynamic changes in lymphocyte subsets and parallel cytokine levels in patients with severe and critical COVID-19. BMC Infect Dis. 2021;21(1):79.
- Yao C, Bora SA, Parimon T, et al. Cell-type-specific immune dysregulation in severely ill COVID-19 patients. Cell Rep. 2021;34(1):108590.
- Carrillo J, Izquierdo-Useros N, Ávila-Nieto C, et al. Humoral immune responses and neutralizing antibodies against SARS-CoV-2; implications in pathogenesis and protective immunity. Biochem Biophys Res Commun. 2021;538:187–191.
- Gaebler C, Wang Z, Lorenzi JCC, et al. Evolution of antibody immunity to SARS-CoV-2. Nature. 2021;591(7851):639–644.
- Liu L, Wang P, Nair MS, et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature. 2020;584(7821):450–456.
- Wu Y, Wang F, Shen C, et al. A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science (80-). 2020;368(6496):1274–1278.
- Sterlin D, Mathian A, Miyara M, et al. IgA dominates the early neutralizing antibody response to SARS-CoV-2. Sci Transl Med. 2021;13(577):eabd2223.
- Russell MW, Moldoveanu Z, Ogra PL, et al. Mucosal immunity in COVID-19: a neglected but critical aspect of SARS-CoV-2 infection. Front Immunol. 2020;11:611337.
- Xu X, Nie S, Wang Y, et al. Dynamics of neutralizing antibody responses to SARS-CoV-2 in patients with COVID-19: an observational study. Signal Transduct Target Ther. 2021;6(1):197.
- Orner EP, Rodgers MA, Hock K, et al. Comparison of SARS-CoV-2 IgM and IgG seroconversion profiles among hospitalized patients in two US cities. Diagn Microbiol Infect Dis. 2021;99(4):115300.
- Fu Y, Li Y, Guo E, et al. Dynamics and correlation among viral positivity, seroconversion, and disease severity in COVID-19: a retrospective study. Ann Intern Med. 2021;174(4):453–461.
- Ng DL, Goldgof GM, Shy BR, et al. SARS-CoV-2 seroprevalence and neutralizing activity in donor and patient blood. Nat Commun. 2020;11(1):4698.
- Lou B, Li TD, Zheng SF, et al. Serology characteristics of SARS-CoV-2 infection since exposure and post symptom onset. Eur Respir J. 2020;56(2):2000763.
- Denning DW, Kilcoyne A, Ucer C. Non-infectious status indicated by detectable IgG antibody to SARS-CoV-2 david. Br Dent J. 2020;229(8):521–524.
- Zaki N, Mohamed EA. The estimations of the COVID-19 incubation period: a scoping reviews of the literature. J Infect Public Health. 2021;14(5):638–646.
- Dispinseri S, Secchi M, Pirillo MF, et al. Neutralizing antibody responses to SARS-CoV-2 in symptomatic COVID-19 is persistent and critical for survival. Nat Commun. 2021;12(1):2670.
- Cohen KW, Linderman SL, Moodie Z, et al. Longitudinal analysis shows durable and broad immune memory after SARS-CoV-2 infection with persisting antibody responses and memory B and T cells. Cell Reports Med. 2021;2(7):100354.
- Wisnivesky JP, Stone K, Bagiella E, et al. Long-term persistence of neutralizing antibodies to SARS-CoV-2 following infection. J Gen Intern Med. 2021;36(10):3289–3291.
- Xiang T, Liang B, Fang Y, et al. Declining levels of neutralizing antibodies against SARS-CoV-2 in convalescent COVID-19 patients one year post symptom onset. Front Immunol. 2021;12:708523.
- Li C, Yu D, Wu X, et al. Twelve-month specific IgG response to SARS-CoV-2 receptor-binding domain among COVID-19 convalescent plasma donors in Wuhan. Nat Commun. 2021;12(1):4144.
- Turner JS, Kim W, Kalaidina E, et al. SARS-CoV-2 infection induces long-lived bone marrow plasma cells in humans. Nature. 2021;595(7867):421–425.
- Kim B, Kim TH. Fundamental role of dendritic cells in inducing Th2 responses. Korean J Intern Med. 2018;33(3):483–489.
- Saravia J, Chapman NM, Chi H. Helper T cell differentiation. Cell Mol Immunol. 2019;16(7):634–643.
- Parackova Z, Bloomfield M, Klocperk A, et al. Neutrophils mediate Th17 promotion in COVID-19 patients. J Leukoc Biol. 2021;109(1):73–76.
- Dorward DA, Russell CD, Um IH, et al. Tissue-specific immunopathology in fatal COVID-19. Am J Respir Crit Care Med. 2021;203(2):192–201.
- Hu B, Huang S, Yin L. The cytokine storm and COVID-19. J Med Virol. 2021;93(1):250–256.
- Mulchandani R, Lyngdoh T, Kakkar AK. Deciphering the COVID-19 cytokine storm: systematic review and meta-analysis. Eur J Clin Invest. 2021;51(1):e13429.
- Chen G, Wu D, Guo W, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. 2020;130(5):2620–2629.
- Voicu S, Ketfi C, Stépanian A, et al. Pathophysiological processes underlying the high prevalence of deep vein thrombosis in critically ill COVID-19 patients. Front Physiol. 2021;11:608788.
- Sagnelli C, Celia B, Monari C, et al. Management of SARS-CoV-2 pneumonia. J Med Virol. 2021;93(3):1276–1287.
- Lee W, Ahn JH, Park HH, et al. COVID-19-activated SREBP2 disturbs cholesterol biosynthesis and leads to cytokine storm. Signal Transduct Target Ther. 2020;5(1):186.
- Sohrabi Y, Reinecke H, Godfrey R. Altered cholesterol and lipid synthesis mediates hyperinflammation in COVID-19. Trends Endocrinol Metab. 2021;32(3):132–134.
- Remy KE, Mazer M, Striker DA, et al. Severe immunosuppression and not a cytokine storm characterizes COVID-19 infections. JCI Insight. 2020;5(17):e140329.
- Leisman DE, Ronner L, Pinotti R, et al. Cytokine elevation in severe and critical COVID-19: a rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir Med. 2020;8(12):1233–1244.
- Trump S, Lukassen S, Anker MS, et al. Hypertension delays viral clearance and exacerbates airway hyperinflammation in patients with COVID-19. Nat Biotechnol. 2021;39(6):705–716.
- Demeulemeester F, de Punder K, van Heijningen M, et al. Obesity as a risk factor for severe COVID-19 and complications: a Review. Cells. 2021;10(4):933.
- Asselah T, Durantel D, Pasmant E, et al. COVID-19: discovery, diagnostics and drug development. J Hepatol. 2021;74(1):168–184.
- Yin S, Huang M, Li D, et al. Difference of coagulation features between severe pneumonia induced by SARS-CoV2 and non-SARS-CoV2. J Thromb Thrombolysis. 2021;51(4):1107–1110.
- Helms J, Tacquard C, Severac F, CRICS TRIGGERSEP Group (Clinical Research in Intensive Care and Sepsis Trial Group for Global Evaluation and Research in Sepsis), et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med. 2020;46(6):1089–1098.
- Fraissé M, Logre E, Pajot O, et al. Thrombotic and hemorrhagic events in critically ill COVID-19 patients: a French Monocenter retrospective study. Crit Care. 2020;24:275. DOI:10.1186/s13054-020-03025-y
- Nahum J, Morichau-Beauchant T, Daviaud F, et al. Venous thrombosis among critically ill patients with coronavirus disease 2019 (COVID-19). JAMA Netw Open. 2020;3(5):e2010478.
- Ren B, Yan F, Deng Z, et al. Extremely high incidence of lower extremity deep venous thrombosis in 48 patients with severe COVID-19 in Wuhan. Circulation. 2020;142(2):181–183.
- Obi AT, Barnes GD, Napolitano LM, et al. Venous thrombosis epidemiology, pathophysiology, and anticoagulant therapies and trials in severe acute respiratory syndrome coronavirus 2 infection. J Vasc Surg Venous Lymphat Disord. 2021;9(1):23–35.
- Iba T, Levy JH, Connors JM, et al. The unique characteristics of COVID-19 coagulopathy. Crit Care. 2020;24:360. DOI:10.1186/s13054-020-03077-0
- Mazzeffi MA, Chow JH, Tanaka K. COVID-19 associated hypercoagulability: manifestations, mechanisms, and management. Shock. 2021;55(4):465–471.
- Barnes BJ, Adrover JM, Baxter-Stoltzfus A, et al. Targeting potential drivers of COVID-19: neutrophil extracellular traps. J Exp Med. 2020;217(6):e20200652.
- Wool GD, Miller JL. The impact of COVID-19 disease on platelets and coagulation. Pathobiology. 2021;88(1):15–27.
- Thachil J, Cushman M, Srivastava A. A proposal for staging COVID-19 coagulopathy. Res Pract Thromb Haemost. 2020;4(5):731–736.
- Zafer MM, El-Mahallawy HA, Ashour HM. Severe COVID-19 and sepsis: Immune pathogenesis and laboratory markers. Microorganisms. 2021;9(1):159.
- Wang J, Saguner AM, An J, et al. Dysfunctional coagulation in COVID-19: from cell to bedside. Adv Ther. 2020;37(7):3033–3039.
- Rosell A, Havervall S, Meijenfeldt V, et al. Patients with COVID-19 have elevated levels of circulating extracellular vesicle tissue factor activity that is associated with severity and mortality-brief report. Arterioscler Thromb Vasc Biol. 2021;41(2):878–882.
- Cañas CA, Cañas F, Bautista-Vargas M, et al. Role of tissue factor in the pathogenesis of COVID-19 and the possible ways to inhibit it. Clin Appl Thromb Hemost. 2021;27:107602962110039.
- Perico L, Benigni A, Casiraghi F, et al. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nat Rev Nephrol. 2021;17(1):46–64.
- Qi X, Keith KA, Huang JH. COVID-19 and stroke: a review. Brain Hemorrhages. 2021;2(2):76–83.
- Ahmadian E, Hosseiniyan Khatibi SM, Razi Soofiyani S, et al. Covid-19 and kidney injury: pathophysiology and molecular mechanisms. Rev Med Virol. 2021;31(3):e2176.
- Kaur S, Bansal R, Kollimuttathuillam S, et al. The looming storm: blood and cytokines in COVID-19. Blood Rev. 2021;46:100743.